



Does Butter Cause Fatty Liver Disease?
The usual suspects have a found a 'new' paper to defend the consumption of seed oils—the Scone Study.

Is it plausible?
Whenever one evaluates a new paper, one has to consider plausibility.
If you read that a paper on homeopathy shows a positive result, well, you have a problem. Because it’s impossible for homeopathic ‘treatments’ to do anything at all, by definition. So putatively positive result is in fact just random variation—even if the P value is less than .05.
Saturated fats cause fatty liver…
So here, when reviewing “Effects of N-6 PUFAs Compared with SFAs on Liver Fat, Lipoproteins, and Inflammation in Abdominal Obesity: A Randomized Controlled Trial” (Bjermo et al., 2012) one must examine the context, and determine if the results are plausible.
Bjermo et al. note:
“Nonalcoholic fatty liver disease (NAFLD)5 affects 25% of the adult population (1) and is strongly associated with metabolic disorders and type 2 diabetes, even independently of abdominal obesity (2, 3).”
However they fail to note that NAFLD is a recent disease. The first modern description of it was in 1980 (Ludwig et al, 1980), almost yesterday in terms of Medicine, and yet in the 32 years between Ludwig and Bjermo, the disease has gone from rare to common.
What changed?
Bjermo and colleagues, who include Ulf Risérus, the senior author, would implicitly have us believe that it’s the massive increase in saturated fat consumption that has occurred over that period, and which apparently continues to this day, as NAFLD rates continue to increase.
“SFAs have been positively related to liver fat (9, 10), whereas the essential [Ω-6] PUFA linoleic acid (18:2n26) has been inversely related to plasma alanine aminotransferase (ALT) concentrations (10).”
So they tested butter vs. “foods rich in [Ω-6] linoleic acid”. Including scones with either “baked-on sunflower oil” or “baked-on butter”. No indication of what the rest of the ingredients were, however.

The participants (one is inclined to say victims), “…were instructed to consume the given food items corresponding to ~15% of energy as linoleic acid.” This is 50% beyond the level considered to by dangerous by the U.S. National Academy of Medicine (IoM):
“An upper boundary for linoleic acid is set at 10 percent of energy for three reasons: (1) individual dietary intakes in the North American population rarely exceed 10 percent of energy, (2) epidemiological evidence for the safety of intakes greater than 10 percent of energy are generally lacking, and (3) high intakes of linoleic acid create a pro-oxidant state that may predispose to several chronic diseases, such as CHD and cancer. Therefore, an AMDR of 5 to 10 percent of energy is estimated for n-6 polyunsaturated fatty acids (linoleic acid).”
But how can SFA cause fatty liver if we are eating less of it?
So is it plausible that the epidemic of fatty liver disease is caused by our eating too much butter and too little saturated fat? See the following two images:


As one can see from the above graphs, our consumption of animal and saturated fat have both been essentially flat since 1980, and the major trend of fat consumption since 1909 has been to increase polyunsaturated vegetable fats like linoleic acid and decrease saturated animal fats.
So no, it’s not plausible.
We know how to cause fatty liver in humans.
Additionally, we do have a standard model for causing fatty liver in humans. It’s an infusion called Intralipid, and it’s comprised of soybean oil, which is mostly linoleic acid. I discussed it at length in this post:
Does Consumption of Omega-6 Seed Oils Worsen ARDS and COVID-19?

1. Sometimes you just get lucky. Several years ago [1.01] I came across the following paper [1.02]: While I have only been able to access the abstract [got it, see PS at end], it's pretty telling of something I've seen implied in a number of different disease processes:
And, despite what the usual suspects claim, it’s the understood primary problem with Intralipid. Note there are other factors that can contribute to what I’d describe as non-pathological fatty liver. Too many calories will result in a short-term increase in fats in the liver, but only Ω-6 fats seem to cause pathogenic fatty liver disease.

There’s also a vast experimental literature describing this process, which I touch on in the post above—even the official guidelines for parenteral nutrition caution against Ω-6 fats. Suffice it to say, it’s a consistent enough effect that the U.S. FDA has approved a treatment for it, which is low in linoleic acid. Tellingly, Intralipid has a similar amount of saturated fat to the non-toxic replacement product, but a far higher level of Ω-6.
Unlike saturated fat or butter over the last many years, soybean oil intake has increased dramatically.

So despite what Bjerno at al., imply, it’s just not plausible that saturated fat consumption and not Ω-6 fats are at fault, from either an epidemiological, clinical, or a mechanistic perspective.
But, fat went up.
But, the fat in the liver did go up during this intervention. What could cause that?
Luckily, Chris Masterjohn did the heavy lifting on this paper back in 2012, “AJCN Publishes A New PUFA Study That Should Make Us Long For the Old Days”, (Masterjohn, 2012).
Chris makes a number of points about this study.
The body preferentially disposes of polyunsaturated fats like linoleic acid.
The primary pathway for this is via oxidation, which means the fats are less likely to be in the liver awaiting storage in adipose tissue. This could explain the short-term divergence.Ω-6 fats preferentially cause liver damage upon oxidative stress.
The fact that these sick people were not grossly ill (they would have been excluded) may have prevented the worst outcomes seen from oxidation of lipids on the liver.Choline allows the liver to export fats, and deficiency results in fatty liver. Protein (which can be used to make choline) is thus required.
Protein consumption went down more in the SFA group, and fat consumption was significantly higher. Fat consumption should have been normalized; choline was not evaluated, or even mentioned.Short-term nature of the experiment.
This is the most important point: other experiments that have found negative effects of Ω-6 fats have typically lasted much longer than this.“Neither diet produced fatty liver.”
This is the most important observation. A short-term intervention like this does not demonstrate that, against the available evidence, saturated fats lead to NAFLD. Additionally, Chris notes that the trends in fat change were not sustainable. If the trend in decline of fat in the liver of the PUFA arm had continued, “…the liver fat of the lucky subjects eating sunflower oil would become so negative that their livers would probably suck their entire bodies into an alternate dimension.”
Force-feeding causes fatty liver.
They made an attempt to keep this intervention weight-neutral. “To avoid weight loss during the intervention, all participants were urged to weigh themselves weekly.” As noted by Nowak, excess calories can cause fat accumulation in the liver, and if the intervention would have caused a decline in calorie intake, ‘making up’ those calories could have increase liver fat. A diet intervention high in saturated fat but geared towards weight loss caused improvement in fatty liver symptoms, over 12 months (Vilar-Gomez et al., 2019).
Did they really measure inflammation?
Bjermo et al., also make some claims about inflammation in their paper, “Fourth, PUFA had no adverse effects on oxidative stress or inflammation…”
Here’s where we run into a problem.
“Dietary Ω-6 PUFA or a high Ω-6/Ω-3 ratio has been suggested to increase inflammation and lipid peroxidation through its conversion to arachidonic acid (20). We found no support for such a hypothesis. Despite the marked increase in linoleic acid intake (14% of energy) and the 3.5-fold increase in the dietary Ω-6/Ω-3 ratio, serum arachidonic acid concentrations were not elevated.”
This is not surprising. Arachidonic acid is a very bio-active fat, and levels in the body are tightly regulated, especially in serum. Similarly, they used two prostaglandins derived from arachidonic acid, “…as indicators of oxidative stress and lipid peroxidation…”.
However the average person eating an industrial diet eats several times the amount of linoleic acid as they do arachidonic, and this specific intervention was to increase linoleic acid. Prostaglandins are produced by the body in a regulated manner as a response to injury or inflammation.
They excluded the people most likely to have high baseline inflammation, which could have resulted in higher conversion of arachidonic acid to prostaglandins: “…diagnosed liver disease, type 1 diabetes, history of a serious cardiovascular event, BMI (in kg/m2) >40, excessive alcohol intake…”
Oxidized linoleic acid byproducts, which can be produced in an unregulated manner, are causes of inflammation.
So a more specific test would have been to measure 4-hydroxynonenal (4-HNE) (Poli et al., 2008) which is “…is one of the major aldehydic metabolites of lipid peroxidation and is considered to be one of the most reliable markers of lipid peroxidation…” and is often used as a indicator of liver damage in pathogenic fatty liver. Unlike the measured prostaglandins, it’s produced directly from linoleic acid, in addition to arachidonic acid. 13-HODE, another oxidized linoleic acid metabolite, has also been used.
This would have helped determine if the fatty liver was likely to progress to a pathological state.
For instance, these authors didn’t find arachidonic acid metabolites to be useful in examining the progression of fatty liver (steatosis) to non-alcoholic steatohepatitis (NASH):
“A risk score for NASH (oxNASH [13-HODE—an oxidized linoleic acid/linoleic acid ratio, age, BMI, and AST]) was developed in the initial clinical cohort and shown to have high diagnostic accuracy for NASH versus steatosis in the independent validation cohort. Subjects with elevated oxNASH levels (top tertile) were 9.7-fold (P < 0.0001) more likely to have NASH than those with low levels (bottom tertile). Collectively, these findings support a key role for free radical-mediated linoleic acid oxidation in human NASH and define a risk score, oxNASH, for noninvasive detection of the presence of NASH.” (Feldstein et al., 2010)

Unfortunately, this belies Bjermo et al.’s claim that, “A high Ω-6 PUFA intake does not cause any signs of inflammation or oxidative stress.” They weren’t using adequate markers to determine if that was the case.
The weight of the evidence clearly indicates a “key role” (Feldstein et al., 2010) for linoleic acid in the pathogenesis of fatty liver.
“Of the markers monitored, 9- and 13-HODEs and 9- and 13-oxoODEs, products of free radical-mediated oxidation of linoleic acid (LA), were significantly elevated in patients with nonalcoholic steatohepatitis (NASH), compared with patients with steatosis. A strong correlation was revealed between these oxidation products and liver histopathology (inflammation, fibrosis, and steatosis).”
Other claims that effect credibility.
Bjermo et al. also claim, “Solid evidence indicates that replacing SFAs with PUFAs reduces coronary artery disease (CAD) events (11, 12) and possibly prevents diabetes (13).”
Calling the evidence to replace SFA with Ω-6 PUFA in CAD “solid” is absurd, as it consistently worsens outcomes in RCTs (Ramsden et al., 2016).
Even the best case one can say is that replacing SFA with Ω-6 PUFA for CVD prevention makes “little or no difference” (Hooper et al., 2018). Not exactly “solid”.
NAFLD is often considered a symptom of diabetes. Just as with NAFLD, type 2 diabetes is a formerly-rare disease that now affects over 50% of the U.S. population, if it’s defined broadly. It’s gone up in correlation with seed oil consumption, and the standard experimental way to induce insulin resistance, the hallmark of diabetes, is with the afore-mentioned Intralipid.
My n=1.
Before I ‘fixed’ my diet by removing seed oils, I had fatty liver, as my physician explained to me. This was based on elevated liver enzymes, which, while not a perfect indicator, is commonly used. Against medical advice—although he had an open mind—I changed my diet, reducing seed oils and carbohydrates, and increasing animal fats including butter and other dairy fats dramatically.
It took four years, but my liver enzymes finally normalized on a diet high in butter, which is where I remain today.
Conclusion
So yes, I find this paper a bit hard to take seriously. I’ll keep eating butter.
P.S.
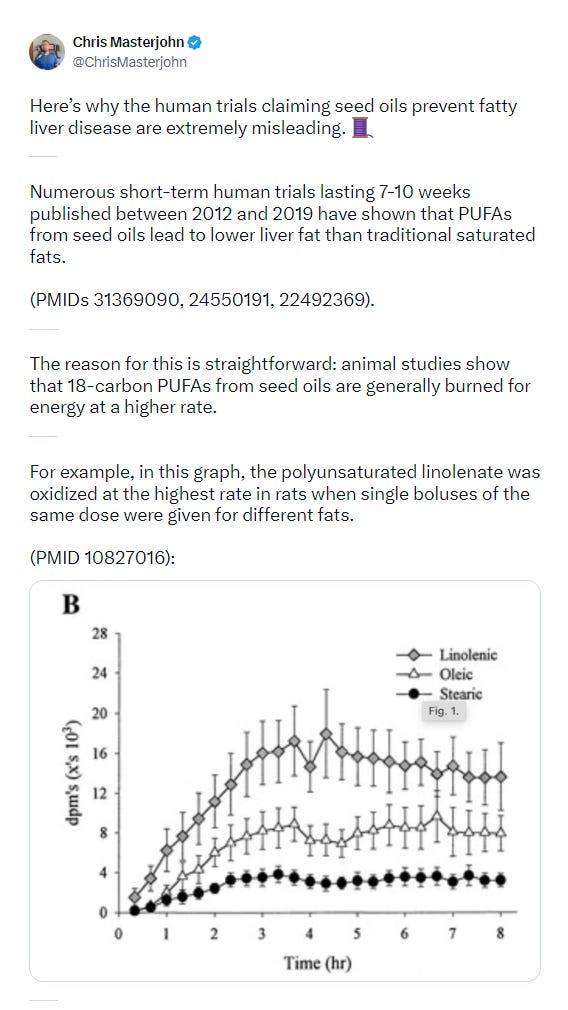
Chris Masterjohn does an updated analysis on X. It’s excellent, do read the whole thing.
References
Bjermo, H., Iggman, D., Kullberg, J., Dahlman, I., Johansson, L., Persson, L., Berglund, J., Pulkki, K., Basu, S., Uusitupa, M., Rudling, M., Arner, P., Cederholm, T., Ahlström, H., & Risérus, U. (2012). Effects of N-6 PUFAs Compared with SFAs on Liver Fat, Lipoproteins, and Inflammation in Abdominal Obesity: A Randomized Controlled Trial. The American Journal of Clinical Nutrition, 95(5), 1003–1012. https://doi.org/10.3945/ajcn.111.030114
Blasbalg, T. L., Hibbeln, J. R., Ramsden, C. E., Majchrzak, S. F., & Rawlings, R. R. (2011). Changes in Consumption of Omega-3 and Omega-6 Fatty Acids in the United States During the 20th Century. The American Journal of Clinical Nutrition, 93(5), 950–962. https://doi.org/10.3945/ajcn.110.006643
Feldstein, A. E., Lopez, R., Tamimi, T. A.-R., Yerian, L., Chung, Y.-M., Berk, M., Zhang, R., McIntyre, T. M., & Hazen, S. L. (2010). Mass spectrometric profiling of oxidized lipid products in human nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Journal of Lipid Research, 51(10), 3046–3054. https://doi.org/10.1194/jlr.M007096
Fresenius Kabi. (08-12/23). Omegaven: Fish Oil Lipid Injectable Emulsion (ILE) [Advertisement]. Fresenius Kabi. https://www.freseniuskabinutrition.com/products/omegaven/
Hooper, L., Al‐Khudairy, L., Abdelhamid, A. S., Rees, K., Brainard, J. S., Brown, T. J., Ajabnoor, S. M., O’Brien, A. T., Winstanley, L. E., Donaldson, D. H., Song, F., & Deane, K. H. (2018). Omega‐6 Fats for the Primary and Secondary Prevention of Cardiovascular Disease. Cochrane Database of Systematic Reviews, 7. https://doi.org/10.1002/14651858.CD011094.pub3
Institute of Medicine. (2005). Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids. https://doi.org/10.17226/10490
Lee, J. H., Duster, M., Roberts, T., & Devinsky, O. (2022). United States Dietary Trends Since 1800: Lack of Association Between Saturated Fatty Acid Consumption and Non-communicable Diseases. Frontiers in Nutrition, 8. https://doi.org/10.3389/fnut.2021.748847
Ludwig, J., Viggiano, T. R., McGill, D. B., & Oh, B. J. (1980). Nonalcoholic Steatohepatitis: Mayo Clinic Experiences with a Hitherto Unnamed Disease. Mayo Clinic Proceedings, 55(7), 434–438.
Masterjohn, C. (2012, May 17). AJCN Publishes A New PUFA Study That Should Make Us Long For the Old Days. The Weston A. Price Foundation. https://www.westonaprice.org/ajcn-publishes-a-new-pufa-study-that-should-make-us-long-for-the-old-days/
Nowak, K. (2020). Parenteral Nutrition–Associated Liver Disease. Clinical Liver Disease, 15(2), 59–62. https://doi.org/10.1002/cld.888
Poli, G., Biasi, F., & Leonarduzzi, G. (2008). 4-Hydroxynonenal–protein adducts: A reliable biomarker of lipid oxidation in liver diseases. Molecular Aspects of Medicine, 29(1), 67–71. https://doi.org/10.1016/j.mam.2007.09.016
Ramsden, C. E., Zamora, D., Majchrzak-Hong, S., Faurot, K. R., Broste, S. K., Frantz, R. P., Davis, J. M., Ringel, A., Suchindran, C. M., & Hibbeln, J. R. (2016). Re-Evaluation of the Traditional Diet-Heart Hypothesis: Analysis of Recovered Data from Minnesota Coronary Experiment (1968-73). BMJ, 353. https://doi.org/10.1136/bmj.i1246
Seki, S., Kitada, T., Yamada, T., Sakaguchi, H., Nakatani, K., & Wakasa, K. (2002). In Situ Detection of Lipid Peroxidation and Oxidative Dna Damage in Non-Alcoholic Fatty Liver Diseases. Journal of Hepatology, 37(1), 56–62. https://doi.org/10.1016/S0168-8278(02)00073-9
Sigma-Aldrich, Inc. (2020, November 11). Intralipid: I141 [Advertisement]. Sigma-Aldrich, Inc. https://www.sigmaaldrich.com/catalog/product/sigma/i141
Vilar-Gomez, E., Athinarayanan, S. J., Adams, R. N., Hallberg, S. J., Bhanpuri, N. H., McKenzie, A. L., Campbell, W. W., McCarter, J. P., Phinney, S. D., Volek, J. S., & Chalasani, N. (2019). Post Hoc Analyses of Surrogate Markers of Non-Alcoholic Fatty Liver Disease (NAFLD) and Liver Fibrosis in Patients with Type 2 Diabetes in a Digitally Supported Continuous Care Intervention: An Open-Label, Non-Randomised Controlled Study. BMJ Open, 9(2), e023597. https://doi.org/10.1136/bmjopen-2018-023597
It needs only an N=1 case to disprove a hypothesis.
"It took four years, but my liver enzymes finally normalized on a diet high in butter, which is where I remain today".
Conclusion: "So yes, I find this paper a bit hard to take seriously. I’ll keep eating butter".
Ditto.
I would respectfully suggest that this "experiment" be repeated by others. Of course P values have a distribution and could vary wildly.