



Does Consumption of Omega-6 Seed Oils Worsen ARDS and COVID-19?
Sometimes you just get lucky. If you want to know why obesity makes COVID-19 outcomes worse, and why Africa had the lowest mortality rates...

1. Sometimes you just get lucky.
Several years ago [1.01] I came across the following paper [1.02]:

While I have only been able to access the abstract [got it, see PS at end], it's pretty telling of something I've seen implied in a number of different disease processes:
"Conclusions: During intensive care treatment, patients with ARDS decrease their percentage plasma concentrations of total plasma linoleic acid, but increase their percentage concentrations of oleic and palmitoleic acids. As plasma linoleic acid concentrations decreased, there was usually an increase in plasma 4-hydroxy-2-nonenal [HNE] values, one of its specific peroxidation products, suggestive of severe oxidative stress leading to molecular damage to lipids." [1.02]
HNE is a highly toxic breakdown product of omega-6 fatty acids (Ω-6), such as the linoleic acid mentioned above, that has been implicated in a number of different disease processes. [1.3] It's also a commonly-used marker of oxidative stress (OxStr), a process in which primarily Ω-6 fats in different cell structures are broken down into various toxins, including HNE. Linoleic acid is the Ω-6 fatty acid which is most commonly consumed in an industrial diet.
This was the first time I'd seen an example of this breakdown happening during the course of a disease, so I made note of it.
2. ARDS is the process by which SARS kills you.
Acute Respiratory Distress Syndrome (ARDS) is how the viruses that cause Severe Acute Respiratory Syndrome (SARS) kill you. SARS is what COVID-19 (AKA SARS2) induces. As one paper, "Is SARS just ARDS?", [2.1] notes:
"Of course SARS is not just ARDS... However, in the ICU [Intensive Care Unit]... SARS is essentially ARDS plus intensified respiratory isolation.... Ninety percent of the critically ill patients were diagnosed with ARDS or acute lung injury. [ALI]"
So as I have an interest in Ω-6 and OxStr, the idea that it may play a part in SARS and COVID-19 was of course of interest to me, too.
Peter Dobromylskyj, author of the esteemed Hyperlipid blog, took the study I tweeted above and started pulling the thread, [2.2] finding this very interesting paper, [2.3] which concludes that:
"Increases in unsaturated serum acyl chain ratios differentiate between healthy and seriously ill patients, and identify those patients likely to develop ARDS. Thus, the serum acyl ratio may not only prospectively identify and facilitate the assessment of new treatments in patients at highest risk for developing ARDS, but may also lead to new insights about the pathogenesis of ARDS." [2.3]
Ω-6 fats, along with omega-3 (Ω-3) fats, are the unsaturated fats that are most bioactive in inflammatory processes in the body.
"The calculated ratios of serum free fatty acids (i.e., the ratio of C18 unsaturated fatty acids [linoleic] and [oleic] to fully saturated palmitate, C16:0) increased and predicted the development of ARDS in at-risk patients." [2.3]
Sadly, again just an abstract.
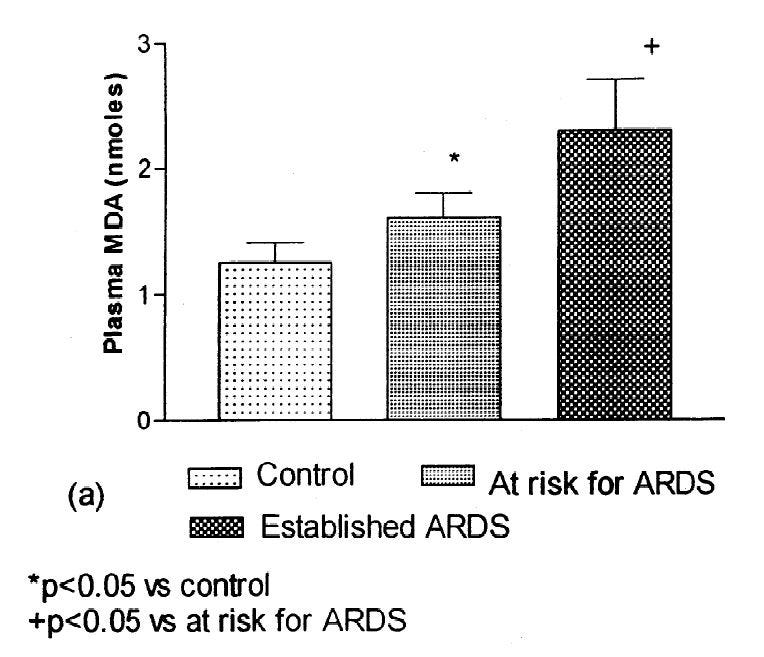
In "Oxidant stress and essential fatty acids in patients with risk and established ARDS", [2.4] they looked at the fatty acid changes in plasma. They found a decrease in Ω-6 polyunsaturated fatty acids (PUFAs), and an increase in monounsaturated (MUFA) and saturated fats (SFA).
Malondialdehyde (MDA), like HNE, is often used to monitor the toxic effects of OxStr, as MDA is in the graph to the left. Elevated MDA or other indicators of OxStr show that PUFAs are breaking down into toxins. Unlike HNE, MDA can come from either Ω-3 or Ω-6 fats, and is thus not a great indicator for a problem with Ω-6 specifically, however it is often used.
This suggests that Ω-6 PUFA is breaking down into its oxidation products, such as MDA, as ARDS commences, although this study was not tracking individuals over time.
3. Do Ω-6 Fats Induce ARDS?
Total Parenteral Nutrition (TPN) is a method by which people who cannot eat via their stomachs are fed; the nutrients are injected via an IV (enteral means via the intestines, parenteral means via something other than the intestines). TPN routinely uses Ω-6 soybean oil as the lipid (fat) energy source, and it's well-recognized that this is extremely problematic [3.1] to the liver. So this got me to wondering: what effect would IV administration of Ω-6 fats have on ARDS?
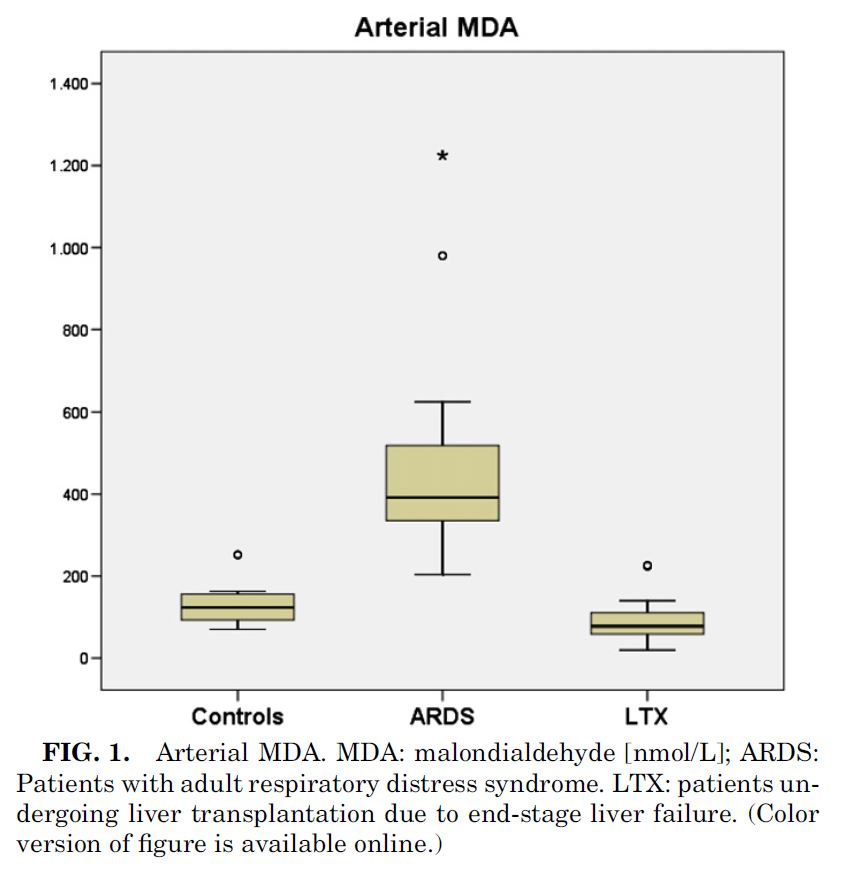
Lichtenstern et. al in 2011 [3.2] looked at the similarities between organ failure of the liver and that induced by ARDS, noting:
"Highly reactive oxygen species generate lipid hydroperoxides by reaction with polyunsaturated fatty acids of cell membranes or plasma lipoproteins that serve as endogenous amplifiers of a complex destructive chain reaction."
Oxidized PUFAs appear to be central to both processes, but as the experience with TPN demonstrates, the Ω-3 fish oil used as an alternative to soybean oil lacks the toxic effects on the liver.
"“Prior to this, all lipid emulsions had a ‘black box’ warning, saying they could cause fat overload syndrome that could be fatal,” says Gura. “We convinced the FDA that Omegaven [fish oil] doesn’t cause this side effect, and we were able to have the black box removed.”" [3.1]
Lichtenstern et. al also looked at MDA to measure lipid peroxidation, even though it's not a great indicator for a problem with Ω-6 specifically. They therefore looked at two other chemicals used to track this process, and noted:
"Hexanal comes up from n–6-PUFA like linoleic and arachidonic acid... In ARDS patients, we found higher concentrations of hexanal than [Ω-3] propanal... ARDS patients show significantly superior hexanal and propanal concentrations in arterial than in mixed venous blood, leading to the conclusion that the lung is the origin of these lipid peroxidation products...."
ARDS can result from a variety of things, which we will discuss later. For the moment, let's discuss trauma. Trauma is the result of injury from some sort of physical cause, like a car accident or a fall. This narrows down the range of things that can cause ARDS, and allows us to focus on the topic at hand.
Sometimes as a result of a traumatic injury a person requires mechanical ventilation; which can require insertion of a tube down the throat. This makes eating difficult, of course, so such people may be put on TPN to provide nourishment while on the ventilator.
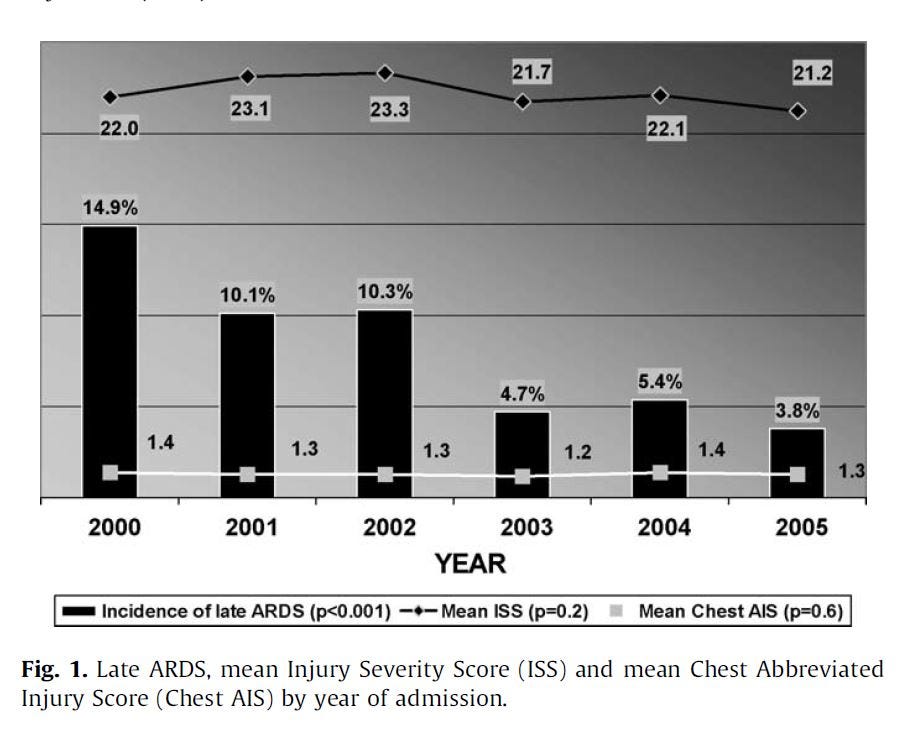
This was not a perfect study, it was not an RCT, but a prospective study of procedures in a level 1 trauma center intensive care unit (ICU). Which means the worst trauma cases were sent there.
They also were using the data they were gathering to improve their practice and better care for their patients as the study went along.
"The incidence of late ARDS among those exposed to TPN was 28.7% (116/404) compared with 3.9% (76/1942) among those not so exposed. Adjustments for potential confounding associated risk factors were made."
So patients put on TPN were 7.4x more likely to get ARDS. That's significant. If this had been an RCT, they likely would have stopped it early for ethical reasons, instead:
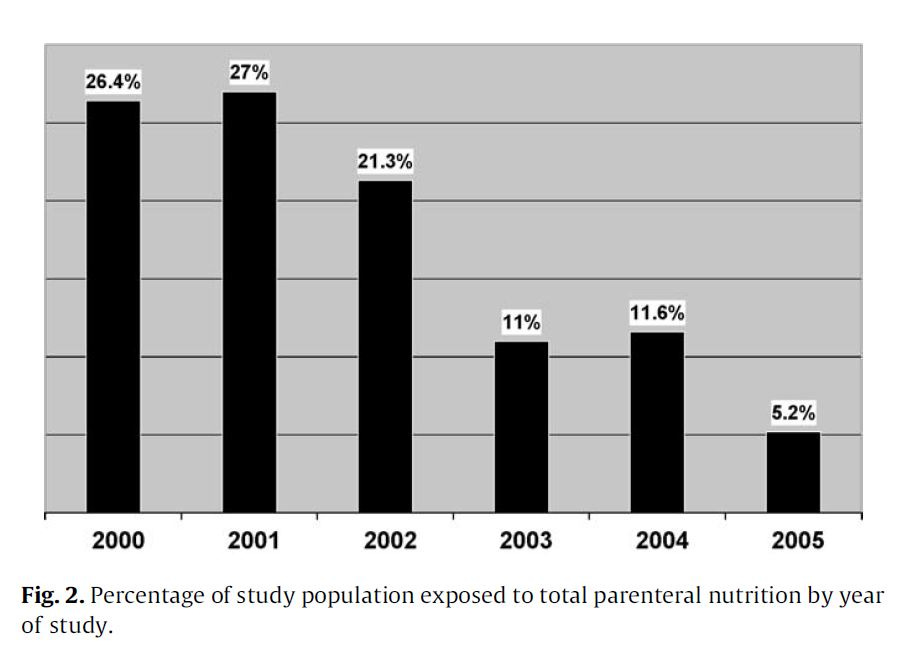
"As shown in Fig. 2, over the study period TPN exposure decreased significantly from 26.4% to 5.2%."
They chose to lower TPN use as the study progressed, and reduced the incidence of an extremely serious condition. Maybe not great science, but good medicine.
They don't specify what they used for TPN, "Exposure to TPN was defined as infusion of a standard formula, including the administration of lipids, before onset of ARDS", but at that time Ω-6 soybean products like Intralipid were standard:
"In patients who are critically ill, commonly used [lipid emulsions]s have provided long-chain triglycerides (LCTs), particularly soybean oil (SO), with a high percentage of ω-6 polyunsaturated fatty acids (PUFAs; 18:2 ω-6) [linoleic acid]." [3.4]
Ω-3 infusions like Omegaven are still not approved for such use. [3.1]
Enteral feeding methods
We know that consumption of linoleic acid versus other PUFAs can dramatically alter the tissue composition of fats, both in animals [3.5, 3.6] and in humans, [3.7, 3.8] and that "dietary omega-6 fatty acid lowering increases bioavailability of omega-3 polyunsaturated fatty acids in human plasma lipid pools", [3.9] so it is plausible that excess consumption or infusion of Ω-6 fats could have a negative effect on lung health, as implied by study [3.3] looking at TPN and ARDS. Papers and meta-analyses that attempt to look at the question of feeding relative amounts of Ω-6 versus Ω-3 fats often don't strictly look at replacing Ω-6 with Ω-3, but instead look at supplementing with Ω-3 [3.4, 3.10, 3.11, 3.12, 3.13, 3.14, 3.15, 3.16], or comparing a low Ω-6 diet to a similar diet supplemented with Ω-3 [3.17]. (They mostly tended to show some benefit, even a meta-analysis looking at safety alone found a mortality benefit. [3.18] — No comments about "statistical significance", please.)
This lack of a direct look at the question no doubt prompted these researchers to compare Ω-6 Intralipid to Ω-3 Omegaven in a rat model of ALI. [3.19]
"Critically ill patients, such as those with acute respiratory distress syndrome (the more severe form of ALI) often require parenteral nutrition, and lipid emulsions are crucial for providing essential fatty acids [7–10], but with the associated risk of causing parenteral nutrition associated liver disease [11–13]."
For a change in studies like this, they actually had a real control—comparing to saline—and prompted ALI with lipopolysaccharides (LPS), a molecular component of bacteria that triggers an immune response without an actual bacterial or viral infection.
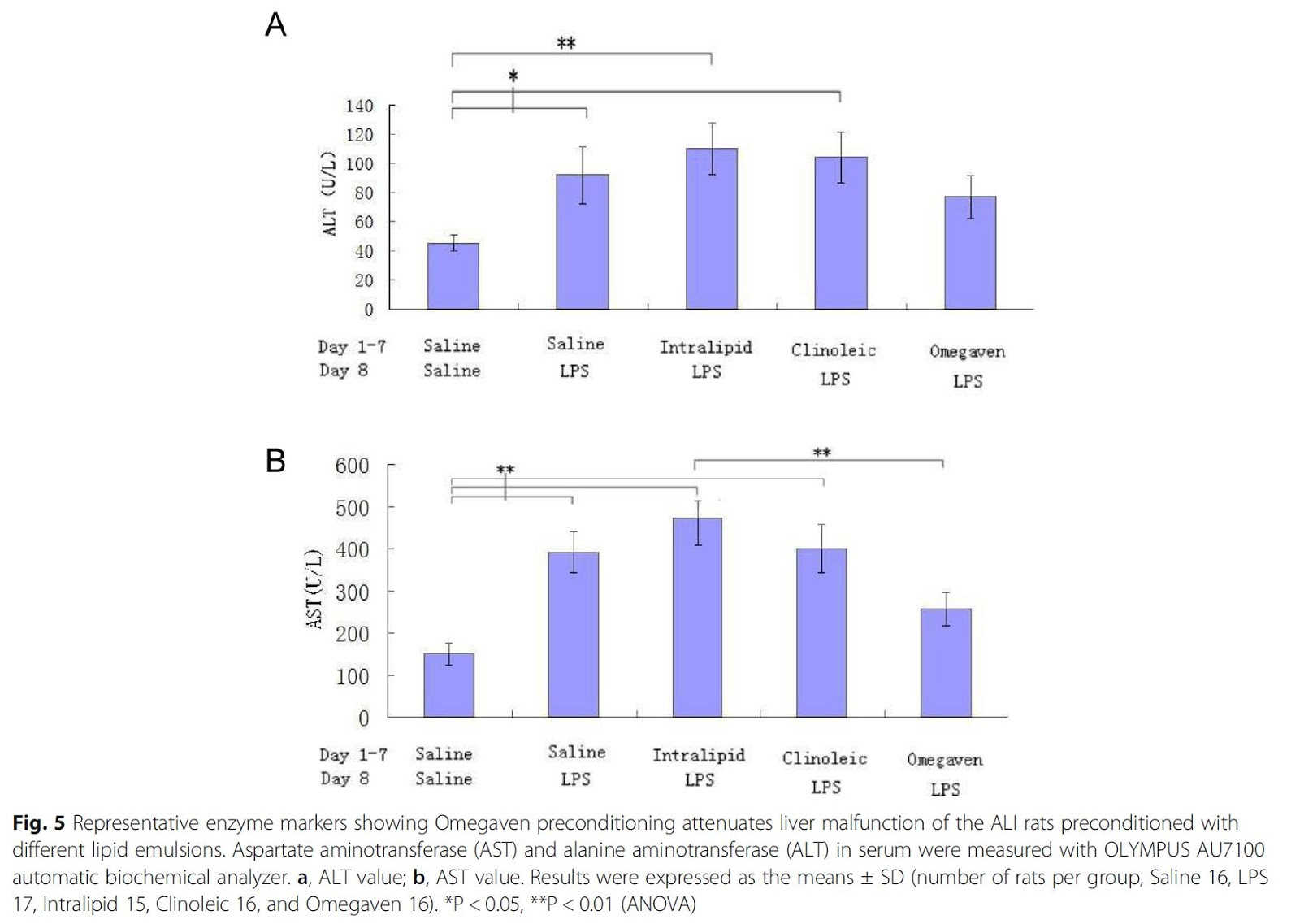
Additionally, they used Clinoleic, an infusion that uses mostly monounsaturated oleic acid, offering a middle ground between the level of Ω-6 in the other two infusions.
Figure 5 (above) shows the expected increase in markers of liver damage from the Ω-6-containing infusions, while the Ω-3 infusions was actually protective for liver damage compared to the control.
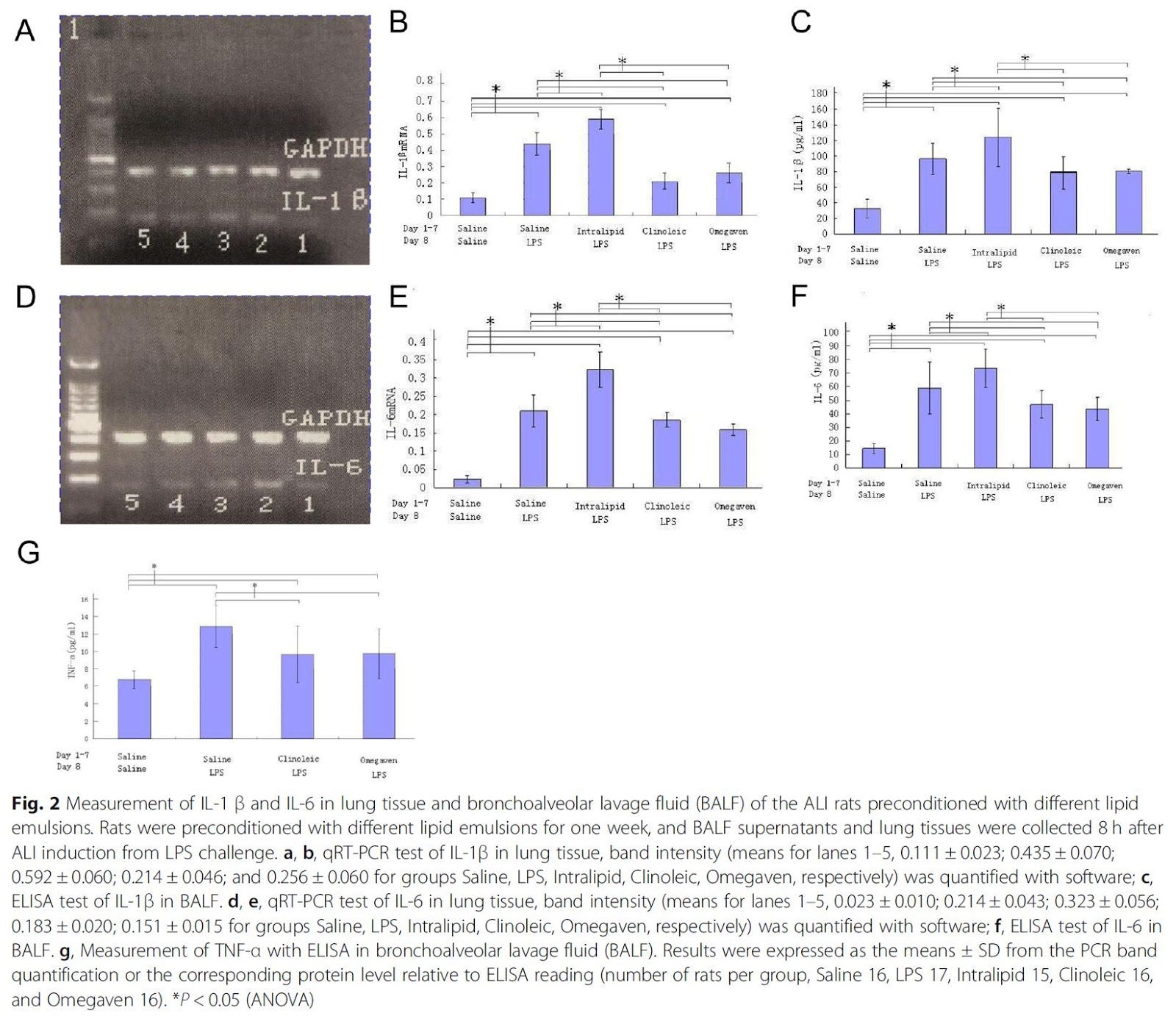
"Although conditions may vary, IL-1β, IL-6 and TNF-α are often considered as the typical early-phase cytokines implicated in the pathogenesis of ALI [24, 25]." [3.19]
Figure 2 (left) shows that the Ω-6 infusion consistently increases levels of inflammatory cytokines versus the other options, often worse than the control while the non-Ω-6 options are better than the saline+LPS control. The only clear exception to this is leukotriene B4 (LTB4), which the Intralipid infusion reduced in these rats. However, Sabater et. al. [3.13], after observing:
"It has also been reported that the higher the plasma levels of LTB4, the higher the degree of hypoxemia. Increased mortality has been observed in patients with higher baseline levels of plasma LTB4 [26,29]. All these data confirm that LTB4 is a proinflammatory molecule with a fundamental role in the cascade that leads to the genesis and evolution of respiratory distress, as well as being an excellent prognostic marker [25,27]." [3.13]
Noted (in humans):
"On administering the lipid emulsions at 0.12 g/kg/h for 12 hours, patients who received the LCT [long-chain triglyceride]-rich emulsion presented increased values in all eicosanoids at the end of the infusion compared with baseline. Twelve hours after the end of the emulsion, TXB2 and 6-keto-PGF1a levels decreased and LTB4 levels, both systemic and pulmonary, continued to increase." [3.13]
Their "LCT-rich emulsion" was the same Intralipid. (Emphasis mine.)
"To summarize, in one of the typical rat ALI settings, preconditioning with Omegaven or Clinoleic was superior to Intralipid in decreasing the intensity of the cytokine storm and apoptosis caused by LPS challenge; in addition, Omegaven had the potential to improve liver function." [3.19]
(Note that 3.19 is using a "preconditioning" of the rats, meaning they're put on the infusion prior to being challenged with LPS. This may or may not be a great model for evaluating TPN infusions, but it's a more realistic model of how people on different diets might be likely to develop ARDS, IMHO. Emphasis mine in the following.)
Shifting guidelines for feeding protocols
Up until 2016, the American guidelines were:
"E2. Patients with ARDS and severe acute lung injury (ALI) should be placed on an enteral formulation characterized by an anti-inflammatory lipid profile (ie, [Ω-3] fish oils, borage oil) and antioxidants. ([Evidence] Grade: A)" [3.20]
However, the new guidelines issued in 2016 say:
"E3. We cannot make a recommendation at this time regarding the routine use of an enteral formulation characterized by an anti-inflammatory lipid profile (eg, omega-3 FOs, borage oil) and antioxidants in patients with ARDS and severe ALI, given conflicting data." [3.21]
After noting that:
"The rationale for pulmonary formulas (high fat to carbohydrate to reduce respiratory quotient) has been shown to be erroneous (effect seen only with overfeeding), and their high content of omega-6 fatty acid may drive inflammatory processes."
They further elaborate that:
"Furthermore, comparison with a commercial formula high in omega-6 fatty acids increased the risk for the effect of a negative control in 2 of the studies."
What? They're rather less clear about this than one would like, but in comparing the 2009 guidelines to the 2016 revision, it seems that two new studies were released that cast doubt on the usefulness of Ω-3 supplements.
One study found that a supplement of Ω-3 fats on top of an unknown diet (how does this stuff pass peer review!?!) had no effect versus a saline control:
"While prior studies of fish oil-containing formulas used an isonitrogenous high-fat formula as a control, we administered enteral saline to our control group. Therefore, it may be that a high-fat control enteral formula in the prior studies was actually harmful, thus allowing the results to appear as though the omega-3 formula was efficacious." [3.22]
What was that control formula? We will get to that. The other study that influenced the guidelines found a rather alarmingly negative effect of Ω-3 supplementation:
"Sixty-day hospital mortality was 26.6% in the n-3 group vs 16.3% in the control group (P=.054), and adjusted 60-day mortality was 25.1% and 17.6% in the n-3 and control groups, respectively (P=.11)....
"This study suggests that twice-daily enteral supplementation of n-3 fatty acids, GLA, and antioxidants change plasma levels of n-3 fatty acids but do not improve clinical outcomes or biomarkers of systemic inflammation in patients with ALI and in fact may be harmful." [3.23]
They also have some thoughts on the difference between their paper and previous papers showing benefit:
"Another major difference between this and previous studies is the composition of the control supplement. The control in the previous studies [12-14] was a commercially available, high-fat formulation containing predominately n-6 and omega-9 (n-9) fatty acids selected to match the percentage of calories from fat, protein, and carbohydrate in the n-3 formulation. In contrast, our study used an isocaloric supplement containing mostly carbohydrate calories instead of lipids. Because n-6 and n-9 fatty acids can be metabolized into inflammatory prostaglandins and series-4 leukotrienes [such as LTB4], [7, 8] it is possible that the control formulations in previous studies may have been proinflammatory, accounting for the differences in outcomes between groups. However, we found no improvement in clinical outcomes for n-3 fatty acid supplementation compared with a largely carbohydrate control."
So they suspect that the "control" food was harmful, and Ω-3 fats only show a benefit in comparison to that harmful control. This explains the comment from the 2016 guidelines paper: "...comparison with a commercial formula high in omega-6 fatty acids increased the risk for the effect of a negative control in 2 of the studies."
A negative control is supposed to have no effect, to be neutral, and in this case they're thinking that the control wasn't actually neutral, but was having a negative effect on the patients, and that fish oil may only be beneficial in comparison. To make it explicit, a member of the team of scientists studying this question observed:
"The three studies that demonstrated a beneficial effect of such supplementation (1–3) used a nonstandard “control” formula, which is high in fats and low in carbohydrates. This formula has approximately 50% of calories resulting from fat compared with approximately 30% fat calories in enteral formulas commonly used in intensive care unit patients. Furthermore, the high-fat control formula consists of a relatively high concentration of n-6 and n-9 fats, which tend to be proinflammatory. Compared with this, a high-fat formula supplemented with anti-inflammatory n-3 fats (and with a higher n-3:n-6 ratio) could show benefit....
"The formula used in each study (Oxepa; Abbott Labs, Abbott Park, IL) is a high-fat/low-carbohydrate product enriched with omega-3 fatty acids (including eicosapentaenoic acid [EPA] and docosahexanoic acid [DHA]), ᵧ-linolenic acid (borage oil), and antioxidant vitamins. In each of the three studies, patients were randomized to receive the enriched formula or an isonitrogenous, isocaloric formula, also high fat/low carbohydrate (Pulmocare; Abbott Labs). Although each study has been criticized for methodologic issues, the aggregate results are quite consistent." [3.24]
Pulmocare is the supposedly neutral control in the papers that formerly informed the guidelines; the ingredients are:
"Ingredients: Liquid Vanilla: Water, Sodium & Calcium Caseinates, Sugar, Canola Oil, Corn Maltodextrin, Medium-chain Triglycerides [MCT], Corn Oil, High Oleic Safflower Oil, [vitamins and minerals]..." [3.25]
So sugars, MCT, and seed oils (canola, corn, and safflower). Very close to the high-fat diet (HFD) we'll discuss.
This may leave you wondering what the guidelines DO recommend for these patients that are on parenteral or enteral nutrition, as they pretty clearly seem to be coming to the conclusion that these Ω-6 fat compositions (soy is discussed here) are harmful:
"Alternative [IV fat emulsions (IVFEs)] may provide outcome benefit over soy-based IVFEs; however, we cannot make a recommendation at this time due to lack of availability of these products in the United States. When these alternative IVFEs (SMOF [soybean oil, MCT, olive oil, and fish oil emulsion], MCT, OO, and FO) become available in the United States, based on expert opinion, we suggest that their use be considered in the critically ill patient who is an appropriate candidate for PN." [3.21]
So we have pretty clear indications that Ω-6 lipids can have toxic effects, in the lungs as they do in the liver, and that one of the mechanisms seem to involve inflammatory cytokines, which are well-recognized as an immune response to oxidized Ω-6 fats. And that these negative effects can be moderated by including Ω-3 fats.
4. Mechanisms for Ω-6 inducement and exacerbation of ARDS: Leukotoxin
So back to Peter at Hyperlipid for this post, "ARDS isoprostanes and isofurans" [4.1] (apparently there's a shortage of commas in the UK to go along with the toilet paper shortages), in which he discusses another paper:
"Finally, to test whether free hemoglobin in human lungs is associated with markers of lipid peroxidation we measured BAL [bronchoalveolar lavage] ispoprostanes and isofurans in sequential BAL aliquots from 5 patients with diffuse alveolar hemorrhage [DAH] (Figure 5F). In patients with alveolar hemorrhage, sequential aliquots of BAL fluid are progressively bloodier. As the aliquot number increased, BAL isoprostane and isofuran levels increased." [4.2]
DAH is an aspect of ARDS, [4.3] although it can be found in other situations.
The two major dietary Ω-6 fats are linoleic and arachidonic acids. These metabolize into countless chemicals in the body, including the afore-mentioned HNE, MDA, and LTB4 [4.3] some toxic, some normal parts of the metabolism; and many, like HNE, are both depending on concentration. Isoprostanes and isofurans are yet more breakdown products. These are not produced by enzymes, which implies that they are ‘messy’, produced in an uncontrolled fashion, either by oxidation by reactive oxygen species generated by mitochondria, catalyzation from iron molecules, or auto-oxidation.
As Peter observes:
"I would just comment that while both isoprostanes and isofurans measured in the study are arachidonic acid non-enzymic derived peroxidation products I can see no reason why the toxic derivatives of linoleic acid peroxidation would not also be formed, though these weren't measured in the study." [4.1]
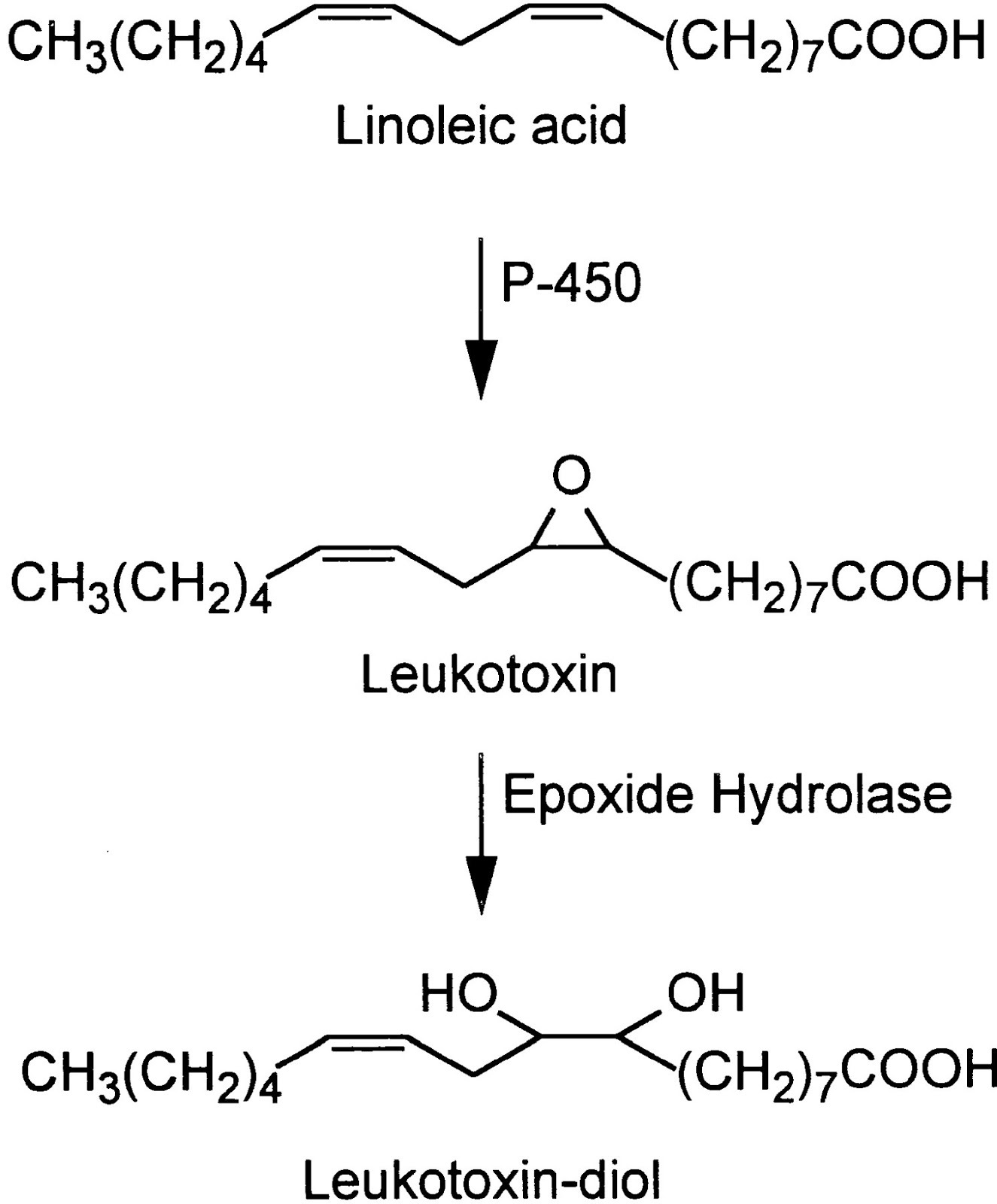
Indeed. In 1986 Japanese researchers found that neutrophils (the most common white blood cells) contained a rather nasty toxin they named leukotoxin (neutrophils are a type of leukocyte, the fancy name for white blood cells). [4.4]
(In an annoying aspect of chemistry, leukotoxins have several different names. Leukotoxin is also known as (±)9(10)-epoxy-12Z-octadecenoic acid and (±)12(13)-epoxy-9Z-octadecenoic acid, or for short, 9(10)-EpOME and 12(13)]-EpOME or just EpOMEs. Leukotoxin-diol, the active toxin, has another series of aliases but is generally referred to as the DiHOMEs. I'll use the common names, but the others may show up in quotes.)
Leukotoxin is a ubiquitous antibiotic and toxin:
"...the gene to manusfect [sic] biosynthesis of [leukotoxin], as a self defensive substance, originates from an early stage of evolution before the separation of animal and plant." [4.4]
As seen in the figure above, leukotoxin is made enzymatically from linoleic acid, the ubiquitous Ω-6 fat, via the cytochrome P450 enzyme, and then in a further step, is made into its highly toxic leukotoxin-diol form by the soluble Epoxyde Hydrolase enzyme (sEH).
"Although the evidence obtained from our previous in vitro studies supported our theory that leukotoxin is a precursor of leukotoxin-diol as a toxic mediator responsible for the development of ARDS, in vivo confirmation was needed....
"...all the mice treated with either leukotoxin-diol or high doses of leukotoxin died of respiratory distress. The symptoms leading ultimately to death were characterized by rapid shallow breathing followed by gasping and discharge of bloody exudate from the nasal and oral cavities. After the appearance of severe respiratory symptoms, the animals given either leukotoxin-diol or leukotoxin (high doses) died in less than 2 min. [4.5]
They also demonstrated that impairing the sEH enzyme reduced mortality, although not from the diol, confirming that the diol is the more toxic element.
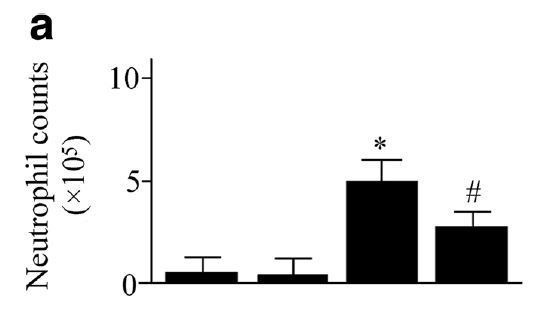
Deletion of the gene responsible for this enzyme [4.6] significantly reduces the harm from hyperoxia-induced lung damage:
"The ROS products were markedly increased in [wild-type] mice exposed to hyperoxia. It was simultaneously associated with an increase in pulmonary inflammation and edema. However, the hyperoxia-induced ROS products and lung injury were dampened in sEH gene knockout mice." [4.6]
Although this authors did not measure leukotoxin, as they did not appear aware of the existing mechanism ("However, the underlying mechanism has not been fully characterized."), it confirms the effect described above.
While the study in [4.5] did not establish the toxin mechanism of leukotoxin-diol, other work has been done:
"Leukotoxin diol specifically activated the mitochondrial permeability transition [MPT], resulting in release of cytochrome c and subsequent cell death. Pretreatment with cyclosporin A inhibited these effects and, furthermore, limited in vivo toxicity." [4.7]
Significantly,
"Because leukotoxin diol, but not leukotoxin, specifically induces ARDS-like symptoms in mice (1), we next examined whether cyclosporin A is an effective inhibitor of leukotoxin diol-induced death and pulmonary edema in this animal model. Mice treated with cyclosporin A prior to leukotoxin diol exhibited dramatically lower mortality than mice that received leukotoxin diol alone (Fig. 5a). Histopathological analysis of murine pulmonary tissue revealed that cyclosporin A pretreatment blocked leukotoxin diol-induced pulmonary hemorrhage and edema, symptoms which are commonly present in ARDS (23) (Fig. 5b). To our knowledge, this is the first time that cyclosporin A has been shown to provide protection against toxic injury to the lung." [4.7]
So a significant negative effect of leukotoxin is due to its ability to kill mitochondria (it's a mitotoxin), thus apparently inducing apoptosis.
"The release of cytochrome c from mitochondria into the cytosol during apoptosis is thought to be dependent on MPT induction (19, 21)."
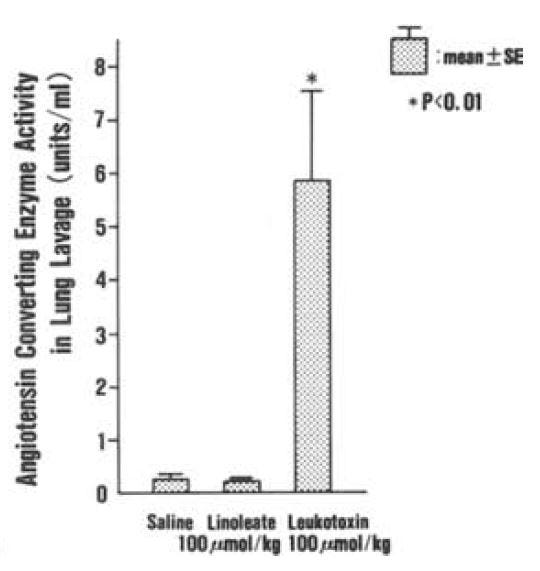
Leukotoxin is found in "considerable" amounts in the lungs of patients suffering from ARDS (and burns, which can also induce ARDS), but not in controls. [4.8] This rat model of ARDS found similar levels of neutrophils, albumin, and angiotensin converting enyzme (ACE) in rats induced to ARDS via hyperoxia (high levels of oxygen). These rats also had leukotoxin as a result (it is presumed) of neutrophil activity, in response to hyperoxia damage. Similar levels of neutrophils, albumin, and ACE could be induced by application of leukotoxin alone, suggesting that leukotoxin is the mechanism by which neutrophils responding to lung infection also induce damage. [4.9] Injection of leukotoxin induces pulmonary edema in rats:
"These morphological findings are similar to those observed in ARDS patients [14]. In addition, the degree of pulmonary edema and increases in albumin concentration and ACE activity in lung lavages are dose- and time-dependent." [4.10]
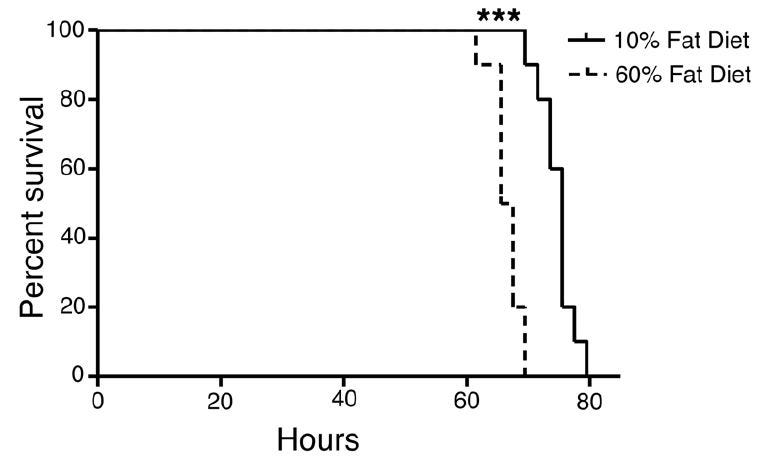
In a mouse model of obesity, [4.11] researchers found that increasing the intake of fat (and Ω-6 fat) increased the plasma levels of leukotoxins, and another group using the same obesity-inducing diet [4.12] found that it increased the susceptibility of mice to the hyperoxic ARDS model discussed in [4.8], although they did not examine levels of leukotoxin. Mice obese through a mechanism that did not depend upon the high-Ω-6 HFD were not susceptible to the enhanced mortality of hyperoxia that affected the HFD-obese mice.
Under certain in vitro conditions, neutrophils have a high capacity to convert linoleic acid to leukotoxin, so it is entirely possible that tissue levels of linoleic acid may influence production of leukotoxin:
"It was noted that about a half ml of linoleate (0.75 mM) added into the leukocyte suspensions (1.6 x 10^7 cells/ml, roughly the same cell counts in blood in patient with bacterial infection) was converted to leukotoxin and its isomer by 10 min incubation at 30°C.... activation of neutrophils either by hypo-osmotic treatment (Table I) or Ca ++ - ionophore (Table II) substantially enhanced the leukotoxin biosynthesis of neutrophils." [4.13]
It's clear that Ω-6 PUFAs are a substrate for leukotoxin production by neutrophils, and also stimulate the release of that toxin.
"A number of polyunsaturated fatty acids stimulate the neutrophil respiratory burst (Badwey et al 1981). Linoleic acid is a potent stimulus of this response; indeed, the response of neutrophils treated with linoleic acid exceeds that observed with arachidonic acid treatment (Li et al 1996)" [4.14]
Exacerbation of ARDS
They're referring to COVID-19; heart damage and coagulation defects have been noted in COVID-19, [4.16] SARS, [4.17, 4.18], and in general ARDS [4.19].
The Japanese researchers who named and have done a lot of the research on leukotoxin note these symptoms as well (here in dogs).
"In our experiments, leukotoxin (9,10-epoxy-12-octadecenoate) depressed cardiac function in a dose dependent manner, and the administration of 50 [mg/kg] of leukotoxin caused severe cardiac dysfunction resulting in death within 45 min. The harmful effects of leukotoxin were much stronger than those of linoleic acid." [4.20]
And:
"These data were compatible with a diagnosis of disseminated intravascular coagulation (DIC).... Over-production of leukotoxin associated with severe infection might therefore account for infection-induced DIC." [4.21]
As leukotoxin is a highly-conserved mechanism of organismal defense, so too are the effects, and they were similar in all animals tested.
A paper observing the lipidomic patterns of severe influenza infections in mice noted:
"The elevated levels of these mediators [leukotoxins] in animals infected with the highly pathogenic influenza strain may explain the severe damage occurring in the lung during infection." [4.22]
There's not a whole lot of research on Ω-6-derived leukotoxin, and there's even less on the cardiovascular effects of it. But it is clearly responsible for some aspects of experimentally-induced ARDS, and it's conceivable that levels of leukotoxin high enough to induce pulmonary failure could also be inducing the other effects here demonstrated only in dogs.
Summary for Leukotoxin
Since we know that diet modulates fatty acid composition of lung tissue ( [3.5, 3.6] ) it is plausible that this increased substrate for leukotoxin production directly affects disease severity in the lung, but this remains to be shown outside the indication given in [4.12]. Given that production of leukotoxin from neutrophils appears to be highly conserved, it is very likely a common mechanism. Certainly leukotoxin is not the only factor contributing to ARDS morbidity and mortality regardless of the cause initiating the process, but given its clear pulmo-toxic effect, it appears to play a major role.
As a final note, many of these models use linoleic acid as a control. While it theoretically can be auto-oxidized to leukotoxin, there is no evidence in the in vivo models I looked at that this happens. It appears to be dependent on neutrophil conversion in response to an injury.
5. Mechanisms for Ω-6 inducement and exacerbation of ARDS: Oxidized Phospholipids (OxPL)
Neutrophils and macrophages (both are leukocytes) have many more roles in organismal defense than as reservoirs and producers of leukotoxin.
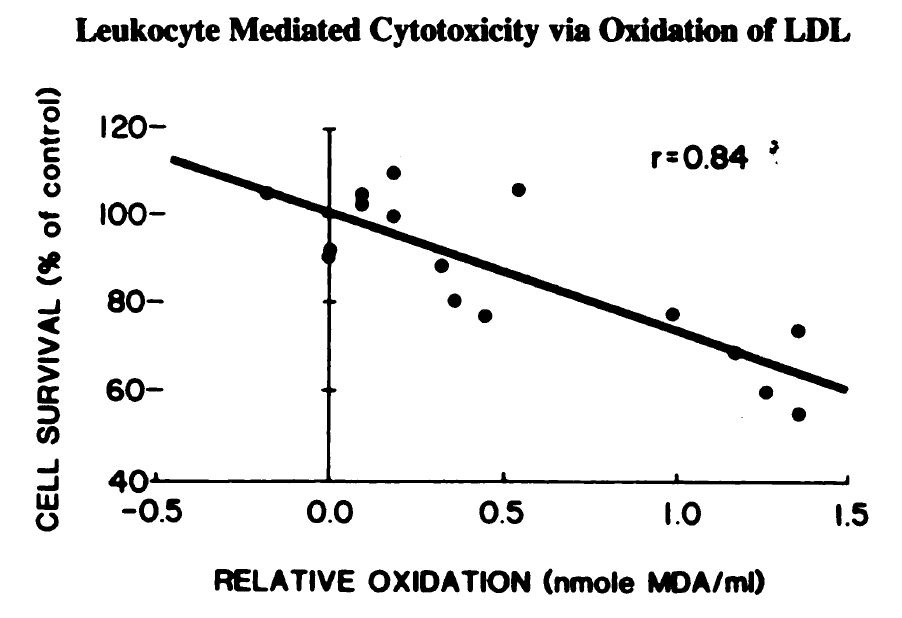
They both have a role in defense via "respiratory bursts":
"Monocytes and neutrophils oxidize low density lipoprotein [LDL] making it cytotoxic" [5.1]
The respiratory burst is another organismal defense mechanism:
"When exposed to certain stimuli, phagocytes (neutrophils, eosinophils, and mononuclear phagocytes) undergo marked changes in the way they handle oxygen (1). Their rates of oxygen uptake increase greatly, sometimes more than 50-fold; they begin to produce large amounts of superoxide (O2-) and hydrogen peroxide (H202); and they start metabolizing large quantities of glucose by way of the hexose monophosphate shunt. Because of the sharp increase in oxygen uptake, this series of changes has come to be known as the "respiratory burst." The "respiration" associated with this burst, however, has nothing to do with energy production; rather, its purpose is to generate powerful microbicidal agents by the partial reduction of oxygen." [5.2]
The demonstration that this burst can cause oxidation of LDL is given here:
"Addition of the free radical scavengers BHT, vit E or GSH virtually prevented
the phagocyte-associated development of TBARS in these experiments and in addition, the subsequent cytotoxicity was not observed. Thus by blocking the free radical mediated monocyte oxidation of LDL the conversion of LDL to a cytotoxin was prevented." [5.1]
"Free radicals" are the things produced by the burst, and absorbing them (what “free radical scavengers” do) prevents the oxidation of LDL into oxidized LDL (oxLDL).
Unsaturated fatty acids, but not saturated, stimulate this respiratory burst in conjunction with tumor necrosis factor (TNF), which is a response to infection, among other things. [5.3] Suppressing TNF reduces OxStr, via a reduction in HNE, in another inflammatory model of organ failure, arthritis. [5.4] So it is very plausible that an unexpectedly high prevalence of PUFA, as exists in the modern Industrial Diet, could exacerbate the creation of oxLDL via the respiratory burst, both by over-stimulation of the burst, and by making phospholipids more susceptible to oxidation.
Now the topic of oxLDL is a large one, and there are many controversies, many of which are outside of the scope of this topic. So I am going to hit a few high points here, and hopefully the references provided will be a jumping-off point for those masochistic enough to pursue the larger topic.
LDL is an instance of a class of molecules used to convey fats and cholesterol around the body. LDL (a "lipo-protein") is comprised of proteins, and lipids in what are called phospholipids (PL). [5.5]
So when one discusses oxLDL, one is discussing oxPL, since what is oxidized in an LDL particle is the PL. However, oxPL is a larger topic than oxLDL, as oxPL includes oxidized phospholipids on cell surfaces and inside cells.
Unlike with leukotoxin, there is clear evidence that oxPL is dependent on dietary intake of fatty acids in humans, and that increased dietary Ω-6 fatty acids predispose LDL to oxidation, [5.6] since it is the amount of Ω-6 linoleic acid in the phospholipids of the LDL or phospholipids in general which makes them susceptible to oxidation [5.8]:
"The results of the present study also show that LDL composition (LDL quality) affects susceptibility to oxidation. The extent of conjugated diene formation in LDL during Cu21-induced oxidation was inversely correlated with the quantity of LDL oleic acid..., and positively correlated with the quantity of LDL linoleic acid... and the 18:2-to-18:1 ratio... ." [5.7]
(Ω-3 fats have a similar susceptibility, but since they are much rarer in the modern, industrial diet, and production of their oxidation products are much lower and less toxic, they have less effect.)
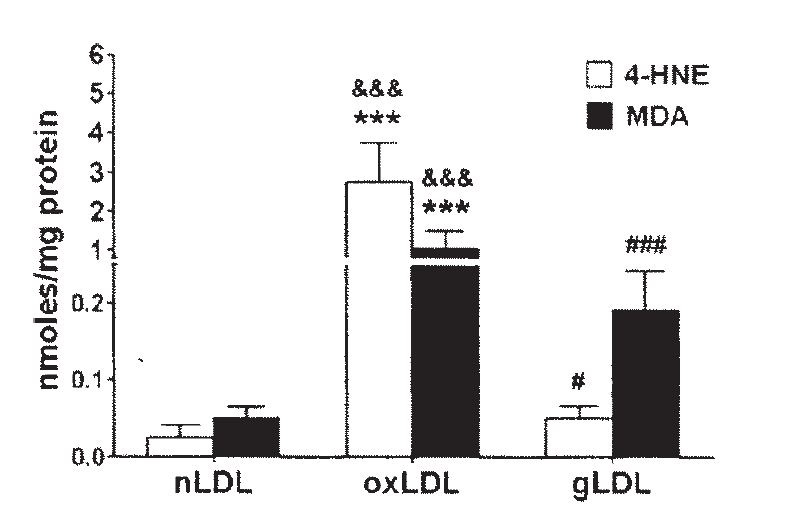
Linoleic acid in oxLDL oxidizes to primarily HNE, but also to some MDA. Ω-6 and the small amount of Ω-3 fats in LDL are almost entirely converted to oxidation products during this process, as saturated fats increase:
"The FA distribution in oxLDL... was altered after LDL oxidation... as compared to the FA distribution in nLDL. The percent of saturated FA in oxLDL increased compared to nLDL: the palmitic acid (C16:0) ... and the stearic acid (C18:0).... The linoleic acid [C18:2] was statistically decreased in oxLDL... compared to nLDL, while the ω-6 dihomogammalinolenic acid (C20:3)... and arachidonic acid (C20:4)... were completely oxidized in oxLDL (fig. 1)." [5.9]
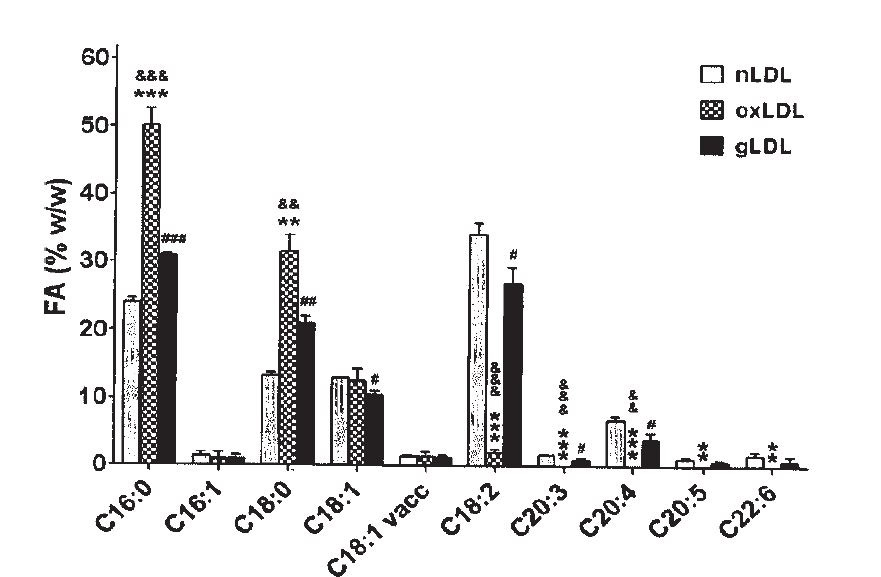
This is, of course, what we saw way back in [1.1], at the beginning of this post. Ω-6 fats converting to toxic metabolites as part of a disease process.
OxPL is Biologically Active and a Self-Antigen
It's not necessary for LDL to be totally oxidized for it to be harmful and part of the disease process. What's known as "minimally modified LDL" is also toxic:
"The effect of minimally modified LDL [mmLDL] on the ability of large vessel endothelial cells (EC) to interact with monocytes and neutrophils was examined. These LDL preparations, obtained by storage or by mild iron oxidation, were indistinguishable from native LDL to the LDL receptor and were not recognized by the scavenger receptor. Treatment of EC with as little as 0.12 µg/ml [mmLDL] caused a significant increase in the production of chemotactic factor for monocytes (sevenfold) and increased monocyte binding (three- to fivefold)." [5.10]
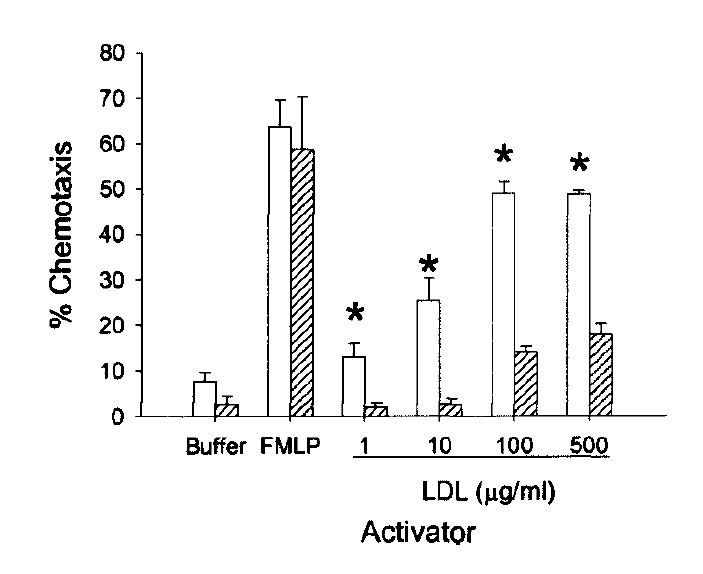
("Chemotaxis" describes how appealing a chemical environment is to a mobile cell. Apparently oxLDL is irresistible to white blood cells...)
Neutrophils are apparently a little more picky, they wait for the oxLDL. [5.11]
Now what's really notable about [5.11] for this discussion, is that they are looking at a model of disease in the lung.
An antigen is a molecule (often a toxin) that induces an immune response in the body. A self-antigen is a molecule that is a normal part of the body that for some reason induces an immune response.
It turns out that oxLDL is both an anti-microbial [5.12] and anti-viral [5.13] toxin, and a self-antigen [5.14]:
"The recognition that oxidative modification of lipoproteins can so fundamentally alter “self” as to elicit an immunologic response underlies our hypothesis that many, if not most, [antibodies against phospholipids] are directed at neoepitopes that are generated as the result of the oxidation of phospholipids." [5.15]
(An "epitope" is the part of a molecule to which an antibody binds.)
This group of scientists developed an antibody test to determine the presence of oxLDL, by binding to the oxidized Ω-6 fats in LDL.
"Although we initially also identified monoclonal antibodies that appeared to bind exclusively to 4-HNE-LDL, these were lost during the cloning procedure." [5.16]
Bummer.
They were able to isolate an antibody, E06*, which is now a standard test for oxPL on oxLDL and other cells, which antibody binds to the oxidized Ω-6 fats in phospholipids:
"E06 did not recognize MDA-LDL, in which 44% of lysines were modified, but did bind to very extensively modified MDA-LDL, presumably due to generation of other oxidation epitopes in these preparations. E06 also bound well to LDL modified by arachidonic acid and, to a lesser extent, LDL modified by linoleic acid oxidation products, and to acrolein-modified LDL. As shown in the companion paper (32), E06 bound prominently to epitopes of oxidized phospholipids and oxidized phospholipid-protein adducts." [5.16]
A later publication from the same group observed:
"Thus, the PC of unoxidized PL, as present in native LDL or viable cells, is “self” and not recognized by innate PRRs. By contrast, the PC of OxPL, present on OxLDL and on apoptotic cells, becomes “altered self” and is recognized by specific [Neutralizing antibodies] NAbs, such as E06; by scavenger receptors [in macrophages, neutrophils, and epithelial cells], such as CD36 and SR-B1; and by innate soluble proteins, such as CRP. In turn, we suggest that each of these PRRs will also recognize the PC present on the cell wall polysaccharide of pathogens, such as S. pneumoniae." [5.17]
This, they observe, is why atherosclerosis and cardiovascular disease is now thought of as an inflammatory disease: it's essentially an autoimmune disease.
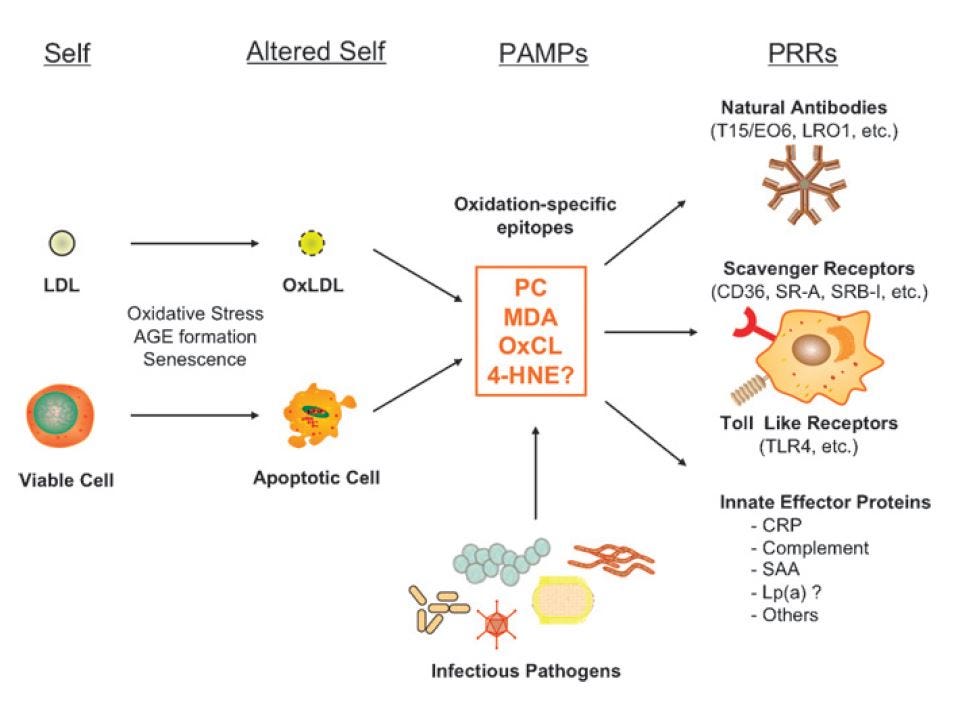
The relevance to ARDS is that much of the damage resulting in mortality appears to be the result of an over-zealous immune response:
"Histological examinations of lung necropsy from SARS patients have demonstrated infiltration of inflammatory cells associated with foamy macrophages, multinuclear syncytial cells and occasional hemophagocytic phagocytes [4], [7]. This has raised the possibility of immunopathological damage of lung tissues due to exaggerated host response but not uncontrolled virus replication in the lung [3]." [5.18]
Macrophages can become "foamy" by stuffing themselves with oxLDL, so it seems pretty clear that the aberrant immune response that can be triggered by over-consumption of Ω-6 fats could be an exacerbating factor.
Protection against oxPL is protective in lung injury
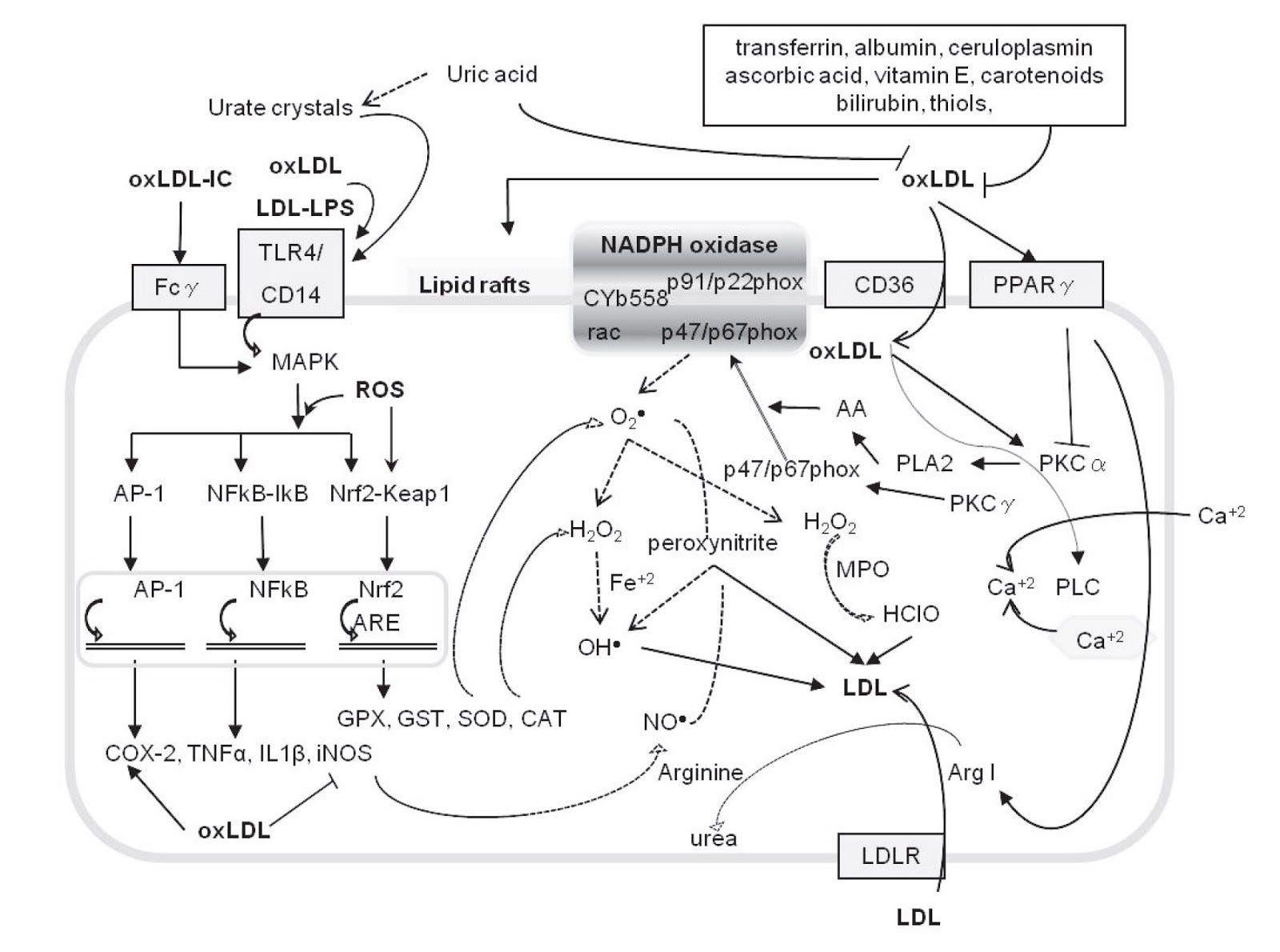
As mentioned above in [5.17], C-reactive protein (CRP) binds to oxPL and to apoptotic cells. Leukotoxin induces apoptosis and oxPL strongly attracts leukocytes that act as if they have encountered an infection when they detect oxPL.
In fact, oxPL induces leukocytes to generate the oxidative burst discussed above.
"It is well known that leukocyte-mediated oxidation of LDL contributes to the pathogenesis of atherosclerosis [1, 3, 4, 6, 7].... Many studies have reported that oxLDL evoke an increase in oxidative burst of leukocytes [71-78]."
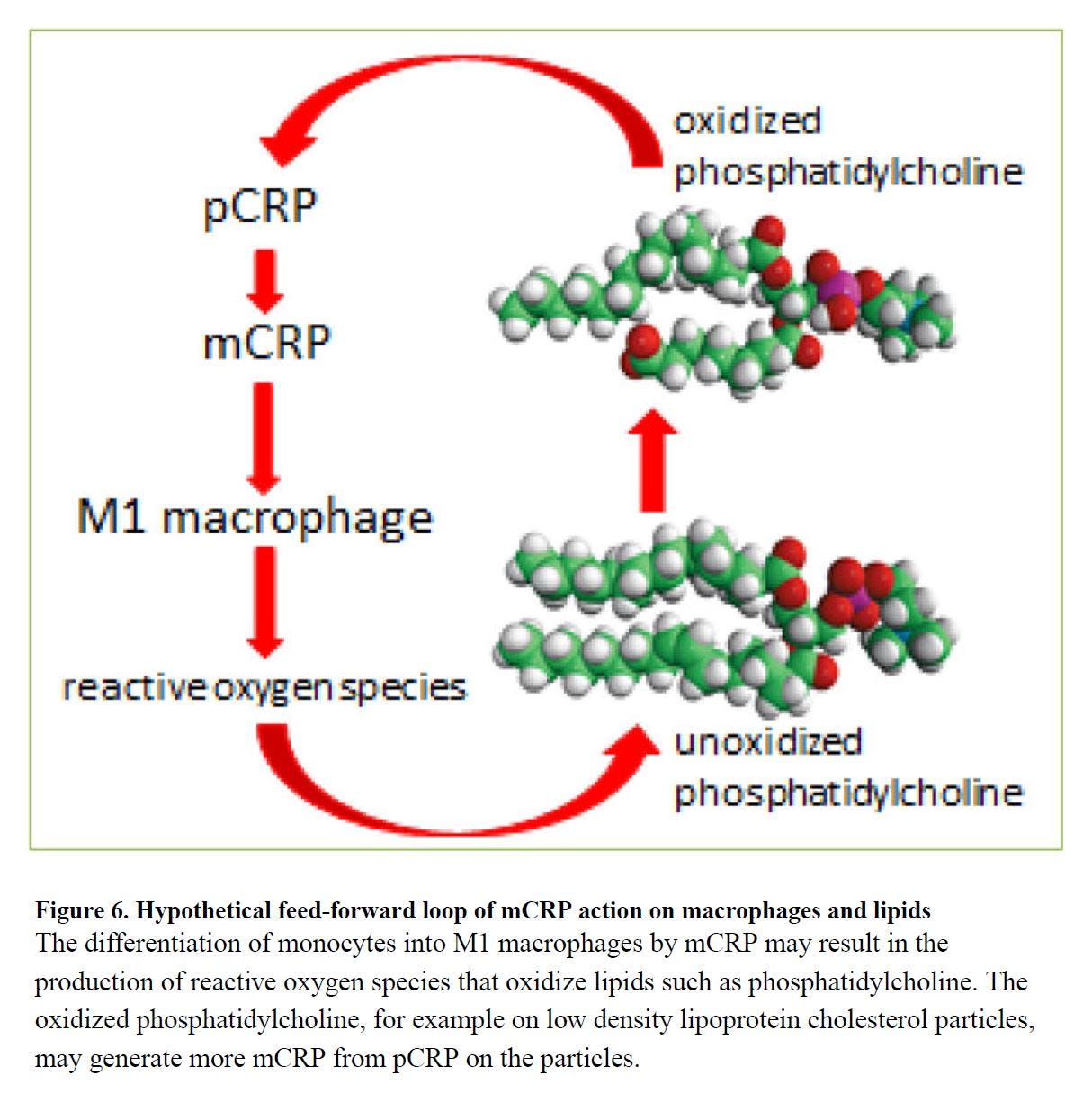
This makes perfect sense if the leukocytes, which aren't the brightest things, interpret oxPL as an infection. Unfortunately, if they execute their burst and find they've made more oxPL, this could easily create a problem where excess neutrophils are attracted to an area trying to end an "infection" which is really just a fire of easily-oxidized fats to which they are adding a spark. [5.19]
You'll note that hypothetical feed-forward loop in [5.19] figure 6 (left) involves CRP. CRP has another useful effect, other than binding to pathogens and things like oxPL that look like pathogens: at high enough levels it begins to suppress the inflammatory response, and in ARDS specifically, this leads to better outcomes:
"We found that among patients with early ARDS, plasma CRP levels were significantly higher among patients who survived 60 days. In keeping with that finding, there was an inverse association between increasing CRP level and 60-day mortality, organ failure, and requirement for mechanical ventilation."
They observe:
"CRP does appear to play an important role in neutrophil chemotaxis, but its role may be more complex than serving to act solely as a chemoattractant; more than 20 years ago, Buchta and colleagues... showed that CRP stimulated chemotaxis at lower concentrations but inhibits it, along with other characteristic neutrophil functions, at higher concentrations. These data appear to establish a basis for our findings that CRP may play a protective role at high concentrations." [5.20]
See also [5.21, 5.22]. This may help explain why higher CRP is protective, as higher neutrophils are associated with worse outcomes in infectious ARDS:
"In patients with ARDS following sepsis, the percentage of BAL polymorphonuclear leukocytes (PMN) was higher on Day 7 (p = 0.11) and particularly Day 14 (p = 0.02) in patients who died; there was a consistent trend of a higher PMN concentration on all days in patients who died then in those who lived." [5.23]
This review, "Accumulating evidence for a role of oxidized phospholipids in infectious diseases", notes:
"In a similar manner, the endogenous acute phase protein C-reactive protein (CRP), which is typically induced during infections such as pneumonia, binds to oxidized phosphorylcholine present in OxLDL and on apoptotic cells [85]. This binding was found to promote the clearance of apoptotic cells and contribute to the resolution of inflammation [86]." [5.24]
So dietary intake of Ω-6 fats influence the fatty acid composition of phospolipids (PL) in the lung, and such modification makes them more susceptible to oxidation, which highly stimulates leukocyte response, which may create a feed-forward loop where responding leukocytes through a respiratory burst response including leukocyte production damage tissue through further oxidation and induction of apoptosis. Such a process is reduced by a high CRP response, which seems to ‘tag’ oxidized fats for clearance, allowing resolution of the potentially damaging neutrophil response.
Reference [5.24] further notes:
"As such it was shown that the antibody EO6,[*] which binds to OxPL on apoptotic cells, has the ability to block inflammation in vitro [25] and in vivo [20]" [5.25]
That's rather interesting. It seems to have rather notable effects in lung models of disease, too:
"Our results in two experimental ALI models, acid aspiration and inactivated H5N1-induced [influenza] ALI, indicate that chemical as well as viral lung pathogens trigger the oxidative stress machinery resulting in ROS generation and the local production of OxPLs...." [5.25]
Moreover:
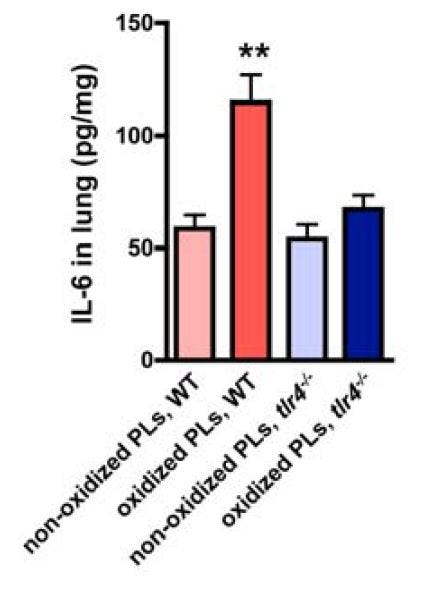
"OxPLs such as OxPAPC can directly trigger cytokine production in macrophages and modulate the severity of acute lung injury in vivo dependent on TLR4-TRIF-TRAF6 expression. Thus, the acute onset of proinflammatory immune responses and severe lung injury caused by different pathogens critically depends on activation of the oxidative stress machinery that couples to innate immunity (Figure S11)." [5.25]
This excessive cytokine production is what's known as the "cytokine storm" that is one of the hazards of a SARS-induced case of ARDS.
They determined the role of oxPL:
"To address whether OxPLs generated during ALI can activate innate immune responses, we stimulated [wild-type (WT)] and tlr4-/- alveolar macrophages with BAL fluid from normal (BAL control) and diseased lungs (BAL acid). BAL fluid from mice with ALI, which contains OxPLs (Figure S5C), induced large amounts of IL-6 production in WT alveolar macrophages. Neutralization of OxPLs by the specific mAb EO6 attenuated BAL fluid-induced IL-6 production (Figure 3E).... Similarly, BAL fluid from diseased mice induced IL-6 production in peritoneal (Figure S6A) and bone marrow macrophages (Figure S6B), which was reverted by EO6 Ab treatment." [5.25]
Indeed, a direct application of oxPL damaged the lungs:
"Importantly, intratracheal administration of OxPAPC induced marked deterioration of lung function in WT mice (Figure 4C)." [5.25]
And in macrophages:
"...IL-6 production in response to surfactant OxPLs was decreased by the neutralizing mAb EO6..." [5.25]
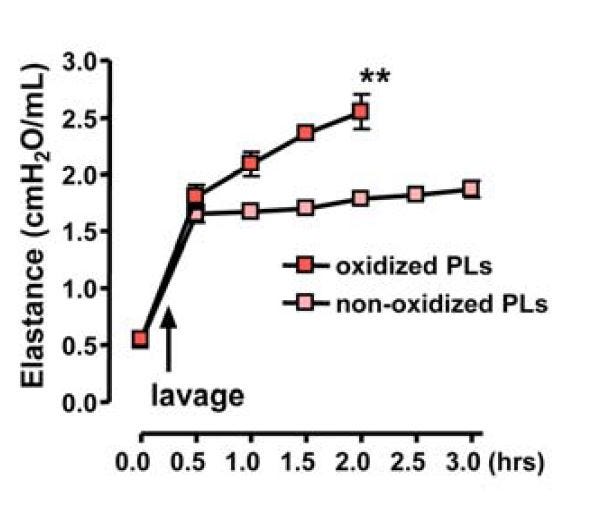
In live mice, they tested replacing normal PL with oxPL:
"In a second experimental model, we removed endogenous surfactant via saline lavages, which strongly impairs lung function; we followed this with intratracheal replacement with nonoxidized or oxidized surfactant PLs. In this scenario, challenge with OxPLs resulted in much more severe impairment of lung function and all experimental animals died by 2 hr, whereas mice that received control, nonmodified surfactant PLs stabilized their lung function (Figure S7A)" [5.25]
Of course, this is in mice. (Emphasis mine.)
"Finally, to test whether OxPLs are generated during actual H5N1 influenza virus infections in humans, we analyzed lung samples from two patients who had developed ARDS following H5N1 avian influenza virus infections. Consistent with our mouse acid aspiration and H5N1 models, we detected massive formation of OxPLs localized at the inflammatory exudates lining the injured air spaces as well as alveolar macrophages in the lungs of both H5N1-infected patients (Figure 7F). Lungs from patients that died of extrapulmonary diseases did not show any detectable OxPL formation (Figure S10C). Thus, H5N1 infections in humans result in the local activation of the oxidative stress machinery and OxPL formation in the lung."
"...Massive formation of OxPLs occurred in all severe cases of acute lung failure we have analyzed including humans who died of H5N1 or SARS infections..."
"...Modulation of this injury pathway, therefore, could be utilized to protect patients infected with H5N1 avian influenza virus, SARS-coronavirus, Anthrax, or other as yet unknown lethal lung pathogens from developing acute severe lung failure." [5.25]
Finally, we have a demonstration that a drug that has undergone human trials can alter lung disease progression (in mice). These authors [5.26] rely heavily on [5.25]:
"In 2008, Imai et al.1 published a highly provocative paper in which they proposed that induction of acute lung injury (ALI), induced by acid aspiration, infection by respiratory viruses and bacteria, or exposure to their products (e.g., inactivated H5N1 influenza), was mediated by a common signaling pathway: NADPH oxidase-dependent production of reactive oxygen species generated oxidized host phospholipids [such as OxPAPC], that, in turn, potently stimulated TLR4." [5.26]
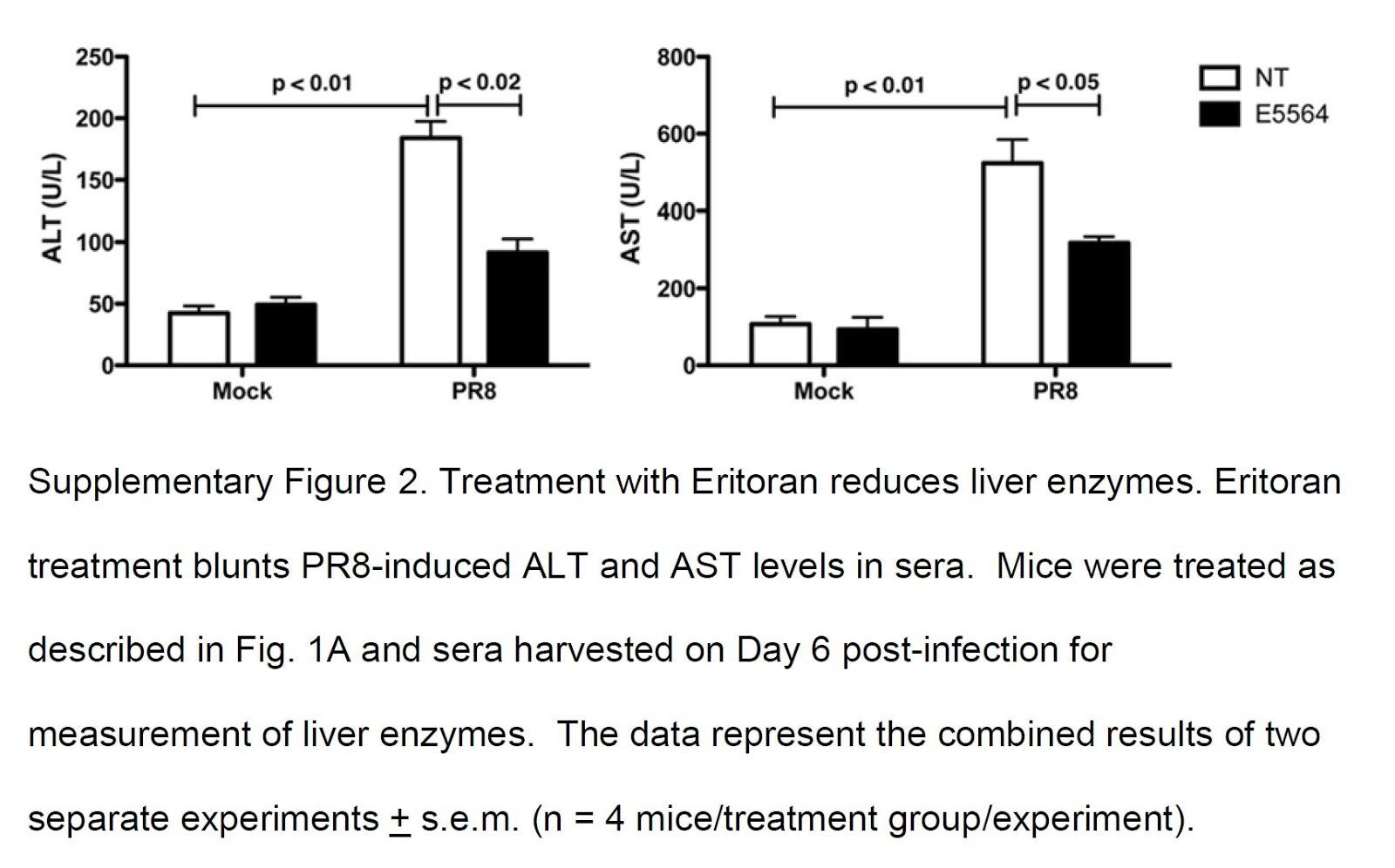
And confirm their results, using a drug, Eritoran, that is a TLR4 blocker (antagonist).
"Together, these results indicate that oxidized phosphatidylcholines [oxPL] are present in influenza-infected lungs and show that treatment with Eritoran reduces the relative abundance of these characteristic ions. Thus, Eritoran treatment of influenza-infected mice not only blocks the cytokine storm exacerbated by influenza-induced endogenous TLR4 agonists, but also inhibits the production and accumulation of certain oxidized phospholipids in infected lungs, including OxPAPC and possibly others." [5.26]
Summary for Oxidized Phospholipids
Here we have a clear pathway from increased dietary Ω-6 fats causing changes in phospholipid composition in humans, making those PLs prone to oxidation, and oxPL playing a significant role in worsened outcomes in various models of infectious and non-infectious lung injury. Addition of oxPL to the lung in surfactant alone will kill experimental mice. This pathway of damage to Ω-6 fats inducing a severe immune response in lung disease seems, as demonstrated in [5.25] and [5.26] seems to be a common pathway underlying ALI regardless of the inciting event. Since virtually all modern human and animal diets contain an evolutionarily novel level of Ω-6 fats versus other types of SFA and MUFA, it seems likely that this has predisposed us to an aberrant inflammatory response.
6. Ω-6 fats are involved in modulating a Cytokine Storm (CS), which worsens ARDS
"Taken together, we have a reasonable hypothesis that cytokine storms play an important role in severe COVID-19 cases. Therefore, neutralising key inflammatory factors in cytokine release syndrome (CRS) will be of great value in reducing mortality in severe cases." [6.1]
CS and cytokines were mentioned several times above, so we will not recapitulate at length the previous topics. To summarize, cytokines are small proteins that have a role in immune system signaling and modulation. Imai 2008 [5.25] observed (to repeat a quotation):
"OxPLs such as OxPAPC can directly trigger cytokine production in macrophages and modulate the severity of acute lung injury in vivo dependent on TLR4-TRIF-TRAF6 expression." [5.25]
Back when we were discussing Ω-6 inducing ARDS, we discussed paper [3.18], which additionally observed:
"To summarize, in one of the typical rat ALI settings, preconditioning with Omegaven or Clinoleic was superior to Intralipid in decreasing the intensity of the cytokine storm and apoptosis caused by LPS challenge; in addition, Omegaven had the potential to improve liver function."
LTB4, discussed above, is involved in cytokine production, and IL-1, IL-6 and TNF-α are all cytokines which are mediators of the immune over-response to oxPL.
Grimble in 1998 ("Nutritional modulation of cytokine biology") observed:
"The ability of peritoneal macrophages from rats to produce IL-1 and IL-6, in response to TNF-a, is greatly influenced by the dietary intake of linoleic acid and total unsaturated fatty acid intake, respectively. IL-1 production increases to plateau concentrations, within a range representing 1–4% of dietary energy, whereas IL-6 production is positively related to unsaturated fatty acid intake over a wider range of intakes." [6.2]
Of course rats aren't people:
"There is a limited amount of evidence that the intake of the [Ω-6] PUFA, LA, intake [sic] may influence pro-inflammatory cytokine production in humans. Monocytes taken from subjects consuming a cholesterol-lowering diet, which involved a reduction in saturated fatty acid intake from 14.1 to 4.0% of dietary energy, and an increase from 6.1 to 8.8% in LA, over a 24-wk period, exhibited enhanced IL-1 and TNF-a production in response to endotoxin. Blood from smokers shows a greater propensity to produce TNF when incubated with endotoxin. It is interesting to note that smokers consuming [Ω-6] PUFA intakes of greater than 5% of dietary energy had two-fold greater concentrations of C-reactive protein than smokers with a lower intake of [Ω-6] PUFAs." [6.2]
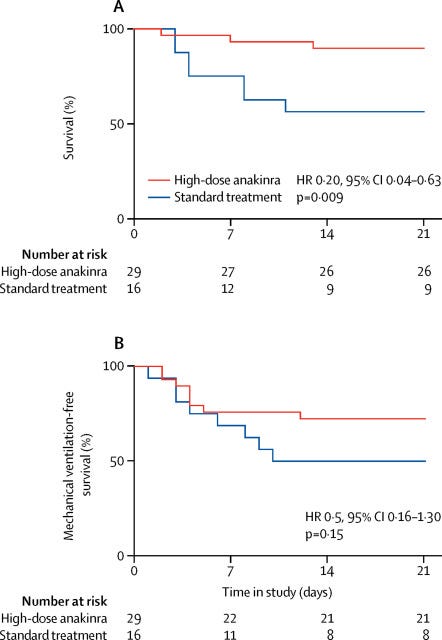
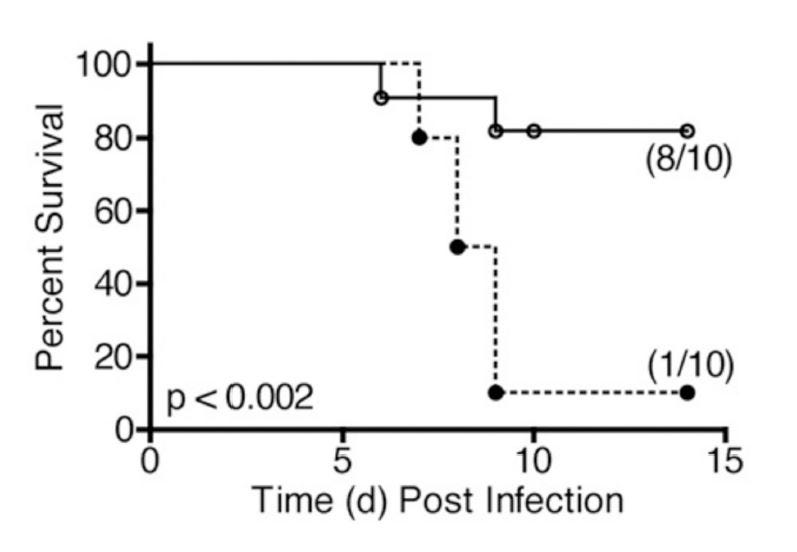
So if increased linoleic acid causes an increase in IL-1, and this has a negative impact on health, then blocking IL-1 might have a benefit. Cavalli et al. (2020) [6.3] examined an "Interleukin-1 [IL-1] blockade with high-dose anakinra in patients with COVID-19, [ARDS], and hyperinflammation: a retrospective cohort study":
"Administration of high-dose intravenous anakinra dampened systemic inflammation and was associated with progressive improvement in respiratory function in patients with COVID-19, moderate-to-severe ARDS, and hyperinflammation, who were managed outside of the ICU in a setting overwhelmed by the COVID-19 pandemic and with a shortage of ICU resources." [6.3]
So if reducing IL-1 can benefit patients with severe ARDS, it seems likely that a dietary intervention to reduce IL-1 may also benefit patients, which could explain why an increased Ω-6 diet seems to be harmful. However, any dietary intervention to reduce linoleic acid to reduce IL-1 or any other cytokine via a reduction in, say, oxPL would take weeks (at best) to alter systemic levels, it is clearly not a useful treatment in an acute-care setting. However, given the massive mortality seen in long-term care settings, where diet is rich in Ω-6 fats—given the dictates of the Dietary Guidelines as enforced by dieticians—it seems that a reconsideration of the impact of high Ω-6 levels on infections needs to be considered.
7. Obesity and Type-2 Diabetes (T2DM) worsen COVID-19 ARDS
Obesity and T2DM, closely-related conditions, both imply a worsened prognosis in COVID-19 ARDS. [7.1, 7.2, 7.3]. As we have seen, a high oxPL status also implies a worse outcome in ARDS, and lowering oxPL can improve ARDS outcomes.
Linoleic acid consumption is also associated with obesity and T2DM, through several mechanisms, including oxPL: [7.4]
"Oxidative stress and inflammation are independently associated with the development of cardiovascular disease ((9),(10),(11),(12),(13),(14)) and [T2DM] in adults ((15),(16),(17),(18),(19),(20)). Oxidized low‐density lipoprotein (oxLDL), a marker of oxidative stress, is associated with obesity ((21),(22)), insulin resistance ((23)), metabolic syndrome ((24),(25),(26),(27)), and cardiovascular disease ((9),(10),(11)) in adults."
A candidate antibody similar to the E06 antibody discussed above also improves insulin resistance (IR), a key attribute of T2DM, in a primate model: [7.5]
"Unexpectedly, we demonstrate that the inhibitory effect of anti-oxLDL on the monocyte-mediated inflammation response is mediated through immune complex formation... We then confirm the anti-inflammatory action of anti-oxLDL in the DIO-NHP model and clearly demonstrate that treatment with anti-oxLDL in DIO-NHP decreased systemic pro-inflammatory markers, improved adaptive immunity and improved insulin sensitivity."
Reduction of linoleic acid also improves obesity and IR in humans [7.6, 7.7, 7.8], and the constellation of symptoms including obesity and IR known as Metabolic Syndrome (MetSyn) is also associated with an inflammatory immune tone, modulated in part by oxPL [7.9, 7.10, 7.11].
It seems reasonable therefore that a high omega-6 diet could worsen the response to a viral infection via worsening of an already pro-inflammatory immune tone, and that the reduction of oxidized omega-6 metabolites including oxPL and leukotoxin via dietary intervention could benefit patients in the longer term.
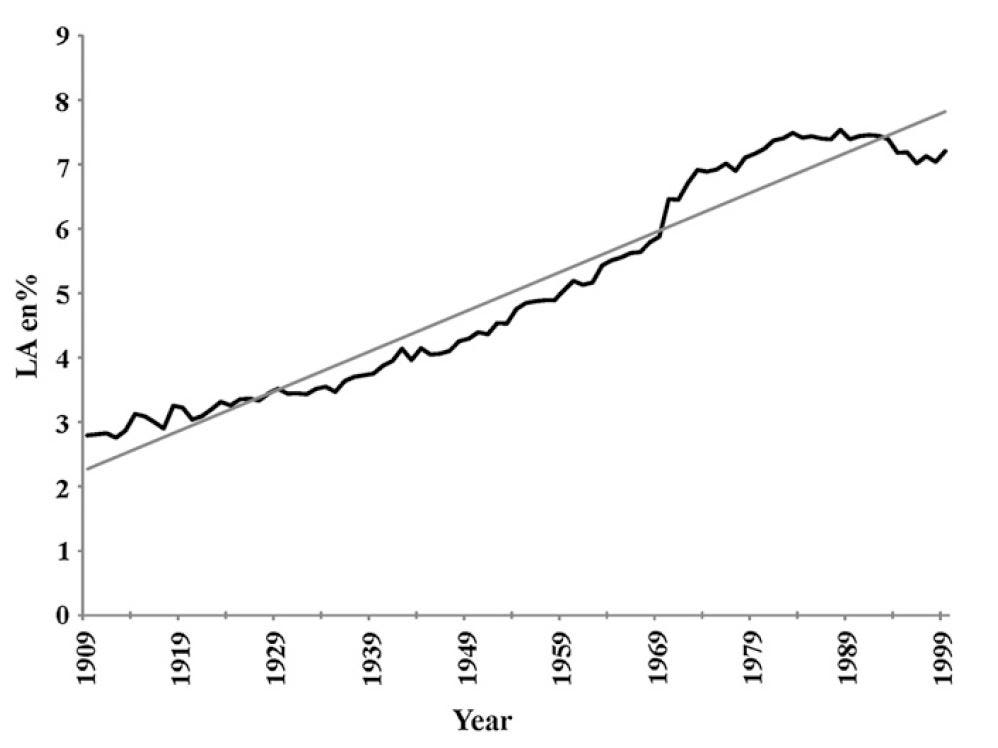
8. Conclusion
The largest single change in the diet [8.1] and composition of the human body [8.2] since 1900 is in the percentage of the omega-6 fat, linoleic acid. This change has been accompanied by epidemics of obesity, T2DM, and cardiovascular disease, indicating a fundamental change has occurred in the functioning of the human body.
The direct negative effect of Ω-6 supplementation in humans and mice on ARDS prognosis alone suggests Ω-6 restriction should be considered as a longer-term prophylactic measure against severe coronavirus infections (as well as for other causes of ARDS), as does the effect of Ω-6 consumption on PL fat composition and oxidation, and the negative effect of that on ARDS.
The clear direct effect of Ω-6-derived leukotoxin on ARDS should warrant significant efforts at determining if the levels of Ω-6 fats which have no precedent in our evolutionary history have altered fundamental immunological pathways in a deleterious fashion.
No dietary change can convey perfect immunity, but as manipulation of dietary factors such as hyperglycemia are an established part of treatment of pulmonary infection [8.3], and as consumption of Ω-6-rich seed oils has a clear effect on pathology of ARDS, it seems that a critical look at this issue is necessary.
For individuals, this should be a further incentive to "fix" their diet to one more in line with our evolution.
Original post dated June 2, 2020.
PS: Thanks to Drs. Toshi Clark and Joseph Mercola for sending the full text of [1.02] to me.
Here's the summary of that paper:
"...This latter finding suggests that peroxidation of linoleic acid seen in the plasma of patients with ARDS probably occurs in the lung, since the lung is undergoing oxidative stress from sequestered neutrophils, and from ventilatory support with high FIO2.
"The data further suggest that providing lipid substrates in the form of enteral or parenteral nutrition to patients experiencing severe oxidative stress may greatly exacerbate the underlying disease process. Specific and sensitive measurements of changes in the plasma polyunsaturated fatty acid linoleic acid, and one of its oxidation products, 4-hydroxy-2-nonenal, support the proposal that patients with established ARDS are under severe oxidative stress from their disease and from treatment with high FIO2 concentrations."
Emphasis mine. The lipid substrate used was Intralipid, discussed above.
* (This antibody is referred to as EO6 and E06 (first with a capital letter O, second with a numeral zero) BY THE SAME AUTHOR in different papers! I'm going to refer to it as E-zero-6 (E06), but will not correct references in quotes.)
References
1.01. Goodrich, Tucker. “Plasma FA changes and incr. lipid peroxidation in patients with ARDS” Showing LA converting to HNE. https://ncbi.nlm.nih.gov/pubmed/8605795 @puddleg. Twitter. Published April 25, 2017.
https://twitter.com/TuckerGoodrich/status/857002343149293569?s=20
1.02. Quinlan GJ, Lamb NJ, Evans TW, Gutteridge JMC. Plasma fatty acid changes and increased lipid peroxidation in patients with adult respiratory distress syndrome. Read Online: Critical Care Medicine | Society of Critical Care Medicine. 1996;24(2):241–246. doi:10.1097/00003246-199602000-00010
1.03. 4-Hydroxynonenal. In: Wikipedia. ; 2020. Accessed April 14, 2020. https://en.wikipedia.org/w/index.php?title=4-Hydroxynonenal&oldid=938910452
2.01. Rubenfeld GD. Is SARS Just ARDS? JAMA. 2003;290(3):397-399. doi:10.1001/jama.290.3.397-a
2.02. Dobromylskyj P. Hyperlipid: ARDS and linoleic acid. Hyperlipid. Published March 17, 2020. Accessed April 14, 2020. https://high-fat-nutrition.blogspot.com/2020/03/ards-and-linoleic-acid.html
2.03. Bursten SL, Federighi DA, Parsons PE, et al. An increase in serum C18 unsaturated free fatty acids as a predictor of the development of acute respiratory distress syndrome. Read Online: Critical Care Medicine | Society of Critical Care Medicine. 1996;24(7):1129–1136. doi:10.1097/00003246-199607000-00011
2.04. Kumar KV, Rao SM, Gayani R, Mohan IK, Naidu MUR. Oxidant stress and essential fatty acids in patients with risk and established ARDS. Clinica Chimica Acta. 2000;298(1):111-120. doi:10.1016/S0009-8981(00)00264-3
3.01. Fliesler N. Omegaven, a life-saving IV nutrition ingredient, clears the FDA - Vector blog. Boston Children’s Hospital Vector. Published August 13, 2018. Accessed April 14, 2020. https://vector.childrenshospital.org/2018/08/omegaven-clears-fda/
3.02. Lichtenstern C, Hofer S, Möllers A, et al. Lipid Peroxidation in Acute Respiratory Distress Syndrome and Liver Failure. Journal of Surgical Research. 2011;168(2):243-252. doi:10.1016/j.jss.2009.10.028
3.03. Plurad D, Green D, Inaba K, Belzberg H, Demetriades D, Rhee P. A 6-year review of total parenteral nutrition use and association with late-onset acute respiratory distress syndrome among ventilated trauma victims. Injury. 2009;40(5):511-515. doi:10.1016/j.injury.2008.07.025
3.04. Manzanares W, Langlois PL, Dhaliwal R, Lemieux M, Heyland DK. Intravenous fish oil lipid emulsions in critically ill patients: an updated systematic review and meta-analysis. Critical Care. 2015;19(1):167. doi:10.1186/s13054-015-0888-7
3.05. Archer S, Nelson D, Gebhard R, Levine AS, Prigge W, Weir EK. Effects of dietary fish oil on lung phospholipid fatty acid composition and intrinsic pulmonary vascular reactivity. Cardiovasc Res. 1987;21(12):928-932. doi:10.1093/cvr/21.12.928
3.06. Baybutt RC, Smith JE, Yeh Y-Y. The effects of dietary fish oil on alveolar type II cell fatty acids and lung surfactant phospholipids. Lipids. 1993;28(3):167-172. doi:10.1007/BF02536635
3.07. Martin MA, Lassek WD, Gaulin SJC, et al. Fatty acid composition in the mature milk of Bolivian forager-horticulturalists: controlled comparisons with a US sample. Matern Child Nutr. 2012;8(3). doi:10.1111/j.1740-8709.2012.00412.x
3.08. Lorenz R, Spengler U, Fischer S, Duhm J, Weber PC. Platelet function, thromboxane formation and blood pressure control during supplementation of the Western diet with cod liver oil. Circulation. 1983;67(3):504-511. doi:10.1161/01.cir.67.3.504
3.09. Taha AY, Cheon Y, Faurot KF, et al. Dietary omega-6 fatty acid lowering increases bioavailability of omega-3 polyunsaturated fatty acids in human plasma lipid pools. Prostaglandins Leukot Essent Fatty Acids. 2014;90(5):151-157. doi:10.1016/j.plefa.2014.02.003
3.10. Mayer K, Grimm H, Grimminger F, Seeger W. Parenteral nutrition with n-3 lipids in sepsis. British Journal of Nutrition. 2002;87(S1):S69-S75. doi:10.1079/BJN2001458
3.11. Chen H, Wang S, Zhao Y, Luo Y, Tong H, Su L. Correlation analysis of omega-3 fatty acids and mortality of sepsis and sepsis-induced ARDS in adults: data from previous randomized controlled trials. Nutr J. 2018;17. doi:10.1186/s12937-018-0356-8
3.12. Langlois PL, D’Aragon F, Hardy G, Manzanares W. Omega-3 polyunsaturated fatty acids in critically ill patients with acute respiratory distress syndrome: A systematic review and meta-analysis. Nutrition. 2019;61:84-92. doi:10.1016/j.nut.2018.10.026
3.13. Gupta A, Govil D, Bhatnagar S, et al. Efficacy and safety of parenteral omega 3 fatty acids in ventilated patients with acute lung injury. Indian J Crit Care Med. 2011;15(2):108-113. doi:10.4103/0972-5229.83019
3.14. Sabater J, Masclans JR, Sacanell J, Chacon P, Sabin P, Planas M. Effects of an omega-3 fatty acid-enriched lipid emulsion on eicosanoid synthesis in acute respiratory distress syndrome (ARDS): A prospective, randomized, double-blind, parallel group study. Nutr Metab (Lond). 2011;8(1):22. doi:10.1186/1743-7075-8-22
3.15. Parish M, Valiyi F, Hamishehkar H, et al. The Effect of Omega-3 Fatty Acids on ARDS: A Randomized Double-Blind Study. Adv Pharm Bull. 2014;4(Suppl 2):555-561. doi:10.5681/apb.2014.082
3.16. (Kristine) Koekkoek W, Panteleon V, van Zanten AR. Current evidence on ω-3 fatty acids in enteral nutrition in the critically ill: A systematic review and meta-analysis. Nutrition. 2019;59:56-68. doi:10.1016/j.nut.2018.07.013
3.17. Sabater J, Masclans JR, Sacanell J, Chacon P, Sabin P, Planas M. Effects on hemodynamics and gas exchange of omega-3 fatty acid-enriched lipid emulsion in acute respiratory distress syndrome (ARDS): a prospective, randomized, double-blind, parallel group study. Lipids Health Dis. 2008;7(1):39. doi:10.1186/1476-511X-7-39
3.18. Chen W, Jiang H, Zhou Z-Y, et al. Is Omega-3 Fatty Acids Enriched Nutrition Support Safe for Critical Ill Patients? A Systematic Review and Meta-Analysis. Nutrients. 2014;6(6):2148-2164. doi:10.3390/nu6062148
3.19. Huang L-M, Hu Q, Huang X, Qian Y, Lai X-H. Preconditioning rats with three lipid emulsions prior to acute lung injury affects cytokine production and cell apoptosis in the lung and liver. Lipids in Health and Disease. 2020;19(1):19. doi:10.1186/s12944-019-1137-x
3.20. McClave SA, Martindale RG, Vanek VW, et al. Guidelines for the Provision and Assessment of Nutrition Support Therapy in the Adult Critically Ill Patient: Journal of Parenteral and Enteral Nutrition. 2009;33(3):277-316. doi:10.1177/0148607109335234
3.21. McClave SA, Taylor BE, Martindale RG, et al. Guidelines for the Provision and Assessment of Nutrition Support Therapy in the Adult Critically Ill Patient. Journal of Parenteral and Enteral Nutrition. 2016;40(2):159-211. doi:10.1177/0148607115621863
3.22. Stapleton RD, Martin TR, Weiss NS, et al. A phase II randomized placebo-controlled trial of omega-3 fatty acids for the treatment of acute lung injury. Crit Care Med. 2011;39(7):1655-1662. doi:10.1097/CCM.0b013e318218669d
3.23. Rice TW, Wheeler AP, Thompson BT, et al. Enteral Omega-3 Fatty Acid, γ-Linolenic Acid, and Antioxidant Supplementation in Acute Lung Injury. JAMA. 2011;306(14):1574-1581. doi:10.1001/jama.2011.1435
3.24. Wiedemann HP. Fish oil is not the fix for acute lung injury*. Read Online: Critical Care Medicine | Society of Critical Care Medicine. 2011;39(7):1829–1830. doi:10.1097/CCM.0b013e31821b82bb
3.25. PulmoCare | Therapeutic Nutrition for People With COPD. Accessed April 22, 2020. https://abbottnutrition.com/pulmocare
4.01. Dobromylskyj, P. (2020, April 14). ARDS isoprostanes and isofurans [Blog]. Hyperlipid. http://high-fat-nutrition.blogspot.com/2020/04/ards-isoprostanes-and-isofurans.html
4.02. Bastarache JA, Sebag SC, Clune JK, et al. Low levels of tissue factor lead to alveolar hemorrhage, potentiating murine acute lung injury and oxidative stress. Thorax. 2012;67(12):1032-1039. doi:10.1136/thoraxjnl-2012-201781
4.03. Alveolar Hemorrhage Syndromes (Diffuse Alveolar Hemorrhage). Pulmonology Advisor. Published January 23, 2019. Accessed April 20, 2020. https://www.pulmonologyadvisor.com/home/decision-support-in-medicine/pulmonary-medicine/alveolar-hemorrhage-syndromes-diffuse-alveolar-hemorrhage/
4.04. Hayakawa M, Sugiyama S, Takamura T, et al. Neutrophils biosynthesize leukotoxin, 9, 10-epoxy-12-octadecenoate. Biochemical and Biophysical Research Communications. 1986;137(1):424-430. doi:10.1016/0006-291X(86)91227-1
4.05. Zheng J, Plopper CG, Lakritz J, Storms DH, Hammock BD. Leukotoxin-Diol: A Putative Toxic Mediator Involved in Acute Respiratory Distress Syndrome. Am J Respir Cell Mol Biol. 2001;25(4):434-438. doi:10.1165/ajrcmb.25.4.4104
4.06. Liu L-P, Li B, Shuai T-K, Zhu L, Li Y-M. Deletion of soluble epoxide hydrolase attenuates mice Hyperoxic acute lung injury. BMC Anesthesiol. 2018;18(1):48. doi:10.1186/s12871-018-0490-z
4.07. Sisemore MF, Zheng J, Yang JC, et al. Cellular Characterization of Leukotoxin Diol-Induced Mitochondrial Dysfunction. Archives of Biochemistry and Biophysics. 2001;392(1):32-37. doi:10.1006/abbi.2001.2434
4.08. Ozawa T, Sugiyama S, Hayakawa M, et al. Existence of Leukotoxin 9,10-Epoxy-12-Octadecenoate in Lung Lavages from Rats Breathing Pure Oxygen and from Patients with the Adult Respiratory Distress Syndrome. Am Rev Respir Dis. 1988;137(3):535-540. doi:10.1164/ajrccm/137.3.535
4.09. Haick AK, Rzepka JP, Brandon E, Balemba OB, Miura TA. Neutrophils are needed for an effective immune response against pulmonary rat coronavirus infection, but also contribute to pathology. J Gen Virol. 2014;95(Pt 3):578-590. doi:10.1099/vir.0.061986-0
4.10. Jia-ning H, Taki F, Sugiyama S, et al. Neutrophil-derived epoxide, 9,10-epoxy-12-octadecenoate, induces pulmonary edema. Lung. 1988;166(1):327-337. doi:10.1007/BF02714065
4.11. Wang W, Yang J, Yang H, et al. Effects of high-fat diet on plasma profiles of eicosanoid metabolites in mice. Prostaglandins & Other Lipid Mediators. 2016;127:9-13. doi:10.1016/j.prostaglandins.2016.11.003
4.12. Plataki M, Fan L, Sanchez E, et al. Fatty acid synthase downregulation contributes to acute lung injury in murine diet-induced obesity. JCI Insight. 2019;4(15). doi:10.1172/jci.insight.127823
4.13. Ozawa T, Sugiyama S, Hayakawa M, Taki F, Hanaki Y. Neutrophil microsomes biosynthesize linoleate epoxide (9, 10-epoxy-12-octadecenoate), A biological active substance. Biochemical and Biophysical Research Communications. 1988;152(3):1310-1318. doi:10.1016/S0006-291X(88)80428-5
4.14. Thompson DA, Hammock BD. Dihydroxyoctadecamonoenoate esters inhibit the neutrophil respiratory burst. J Biosci. 2007;32(2):279-291. Accessed April 27, 2020. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1904342/
4.15. Wadman M, Couzin-Frankel J, Kaiser J, Matacic C, 2020, Pm 6:45. How does coronavirus kill? Clinicians trace a ferocious rampage through the body, from brain to toes. Science | AAAS. Published April 17, 2020. Accessed April 24, 2020. https://www.sciencemag.org/news/2020/04/how-does-coronavirus-kill-clinicians-trace-ferocious-rampage-through-body-brain-toes
4.16. Klok FA, Kruip MJHA, Meer NJM van der, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thrombosis Research. 2020;0(0). doi:10.1016/j.thromres.2020.04.013
4.17. Wu EB, Sung JJ. Haemorrhagic-fever-like changes and normal chest radiograph in a doctor with SARS. The Lancet. 2003;361(9368):1520-1521. doi:10.1016/S0140-6736(03)13170-4
4.18. Chong PY, Chui P, Ling AE, et al. Analysis of Deaths During the Severe Acute Respiratory Syndrome (SARS) Epidemic in Singapore: Challenges in Determining a SARS Diagnosis. Archives of Pathology & Laboratory Medicine. 2004;128(2):195-204. doi:10.1043/1543-2165(2004)128<195:AODDTS>2.0.CO;2
4.19. Abraham E. Coagulation Abnormalities in Acute Lung Injury and Sepsis. Am J Respir Cell Mol Biol. 2000;22(4):401-404. doi:10.1165/ajrcmb.22.4.f184
4.20. Fukushima A, Hayakawa M, Sugiyama. S, et al. Cardiovascular effects of leukotoxin (9,10-epoxy-12-octadecenoate) and free fatty acids in dogs. Cardiovasc Res. 1988;22(3):213-218. doi:10.1093/cvr/22.3.213
4.21. Sugiyama S, Hayakawa M, Hanaki Y, Hieda N, Asai J, Ozawa T. The role of leukotoxin (9, 10-epoxy-12-octadecenoate) in the genesis of coagulation abnormalities. Life Sciences. 1988;43(3):221-227. doi:10.1016/0024-3205(88)90311-6
4.22. Tam VC, Quehenberger O, Oshansky CM, et al. Lipidomic Profiling of Influenza Infection Identifies Mediators that Induce and Resolve Inflammation. Cell. 2013;154(1):213-227. doi:10.1016/j.cell.2013.05.052
5.01. Cathcart MK, Morel DW, Chisolm GM. Monocytes and Neutrophils Oxidize Low Density Lipoprotein Making It Cytotoxic. Journal of Leukocyte Biology. 1985;38(2):341-350. doi:10.1002/jlb.38.2.341
5.02. Babior BM. The respiratory burst of phagocytes. J Clin Invest. 1984;73(3):599-601. Accessed April 24, 2020. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC425058/
5.03. Li Y, Ferrante A, Poulos A, Harvey DP. Neutrophil oxygen radical generation. Synergistic responses to tumor necrosis factor and mono/polyunsaturated fatty acids. J Clin Invest. 1996;97(7):1605-1609. doi:10.1172/JCI118585
5.04. Biniecka M, Kennedy A, Ng CT, et al. Successful tumour necrosis factor (TNF) blocking therapy suppresses oxidative stress and hypoxia-induced mitochondrial mutagenesis in inflammatory arthritis. Arthritis Res Ther. 2011;13(4):R121. doi:10.1186/ar3424
5.05. Dashti M, Kulik W, Hoek F, Veerman EC, Peppelenbosch MP, Rezaee F. A Phospholipidomic Analysis of All Defined Human Plasma Lipoproteins. Scientific Reports. 2011;1(1):1-11. doi:10.1038/srep00139
5.06. Reaven P, Parthasarathy S, Grasse BJ, Miller E, Steinberg D, Witztum JL. Effects of oleate-rich and linoleate-rich diets on the susceptibility of low density lipoprotein to oxidative modification in mildly hypercholesterolemic subjects. J Clin Invest. 1993;91(2):668-676. Accessed February 22, 2019. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC288008/
5.07. Yu-Poth S, Etherton TD, Reddy CC, et al. Lowering Dietary Saturated Fat and Total Fat Reduces the Oxidative Susceptibility of LDL in Healthy Men and Women. J Nutr. 2000;130(9):2228-2237. doi:10.1093/jn/130.9.2228
5.08. Parthasarathy S, Khoo JC, Miller E, Barnett J, Witztum JL, Steinberg D. Low density lipoprotein rich in oleic acid is protected against oxidative modification: implications for dietary prevention of atherosclerosis. PNAS. 1990;87(10):3894-3898. doi:10.1073/pnas.87.10.3894
5.09. Deleanu M, Sanda GM, Stancu CS, Popa ME, Sima AV. Profiles of Fatty Acids and the Main Lipid Peroxidation Products of Human Atherogenic Low Density Lipoproteins. REVCHIM. 2016;67(1):8-12. http://www.revistadechimie.ro/article_eng.asp?ID=4799
5.10. Berliner JA, Territo MC, Sevanian A, et al. Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. J Clin Invest. 1990;85(4):1260-1266. Accessed April 24, 2020. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC296561/
5.11. Sedgwick JB, Hwang YS, Gerbyshak HA, Kita H, Busse WW. Oxidized Low-Density Lipoprotein Activates Migration and Degranulation of Human Granulocytes. Am J Respir Cell Mol Biol. 2003;29(6):702-709. doi:10.1165/rcmb.2002-0257OC
5.12. Hall PR, Elmore BO, Spang CH, et al. Nox2 Modification of LDL Is Essential for Optimal Apolipoprotein B-mediated Control of agr Type III Staphylococcus aureus Quorum-sensing. PLOS Pathogens. 2013;9(2):e1003166. doi:10.1371/journal.ppat.1003166
5.14. Miller YI, Chang M-K, Binder CJ, Shaw PX, Witztum JL. Oxidized low density lipoprotein and innate immune receptors. Current Opinion in Lipidology. 2003;14(5):437–445. Accessed April 13, 2020. https://journals.lww.com/co-lipidology/Abstract/2003/10000/Oxidized_low_density_lipoprotein_and_innate_immune.4.aspx
5.15. Hörkkö S, Miller E, Dudl E, et al. Antiphospholipid antibodies are directed against epitopes of oxidized phospholipids. Recognition of cardiolipin by monoclonal antibodies to epitopes of oxidized low density lipoprotein. J Clin Invest. 1996;98(3):815-825. doi:10.1172/JCI118854
5.16. Palinski W, Hörkkö S, Miller E, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest. 1996;98(3):800-814. doi:10.1172/JCI118853
5.17. Hartvigsen K, Chou M-Y, Hansen LF, et al. The role of innate immunity in atherogenesis. J Lipid Res. 2009;50(Supplement):S388-S393. doi:10.1194/jlr.R800100-JLR200
5.18. Peluso I, Morabito G, Urban L, Serafi FI and M. Oxidative Stress in Atherosclerosis Development: The Central Role of LDL and Oxidative Burst. Endocrine, Metabolic & Immune Disorders - Drug Targets. Published November 30, 2012. Accessed April 29, 2020. http://www.eurekaselect.com/104298/article
5.19. Trial J, Potempa LA, Entman ML. The role of C-reactive protein in innate and acquired inflammation: new perspectives. Inflamm Cell Signal. 2016;3(2). Accessed April 30, 2020. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5058362/
5.20. Bajwa EK, Khan UA, Januzzi JL, Gong MN, Thompson BT, Christiani DC. Plasma C-Reactive Protein Levels Are Associated With Improved Outcome in ARDS. Chest. 2009;136(2):471-480. doi:10.1378/chest.08-2413
5.21. Heuertz RM, Webster RO. Role of C-reactive protein in acute lung injury. Molecular Medicine Today. 1997;3(12):539-545. doi:10.1016/S1357-4310(97)01145-3
5.22. Kew RR, Hyers TM, Webster RO. Human C-reactive protein inhibits neutrophil chemotaxis in vitro: possible implications for the adult respiratory distress syndrome. J Lab Clin Med. 1990;115(3):339-345. https://pubmed.ncbi.nlm.nih.gov/2313163/
5.23. Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med. 1994;150(1):113-122. doi:10.1164/ajrccm.150.1.8025736
5.24. Matt U, Sharif O, Martins R, Knapp S. Accumulating evidence for a role of oxidized phospholipids in infectious diseases. Cell Mol Life Sci. 2015;72(6):1059-1071. doi:10.1007/s00018-014-1780-3
5.25. Imai Y, Kuba K, Neely GG, et al. Identification of Oxidative Stress and Toll-like Receptor 4 Signaling as a Key Pathway of Acute Lung Injury. Cell. 2008;133(2):235-249. doi:10.1016/j.cell.2008.02.043
5.26. Shirey KA, Lai W, Scott AJ, et al. The TLR4 Antagonist, Eritoran, Protects Mice from Lethal Influenza Infection. Nature. 2013;497(7450):498-502. doi:10.1038/nature12118
6.01. Zhang C, Wu Z, Li J-W, Zhao H, Wang G-Q. Cytokine release syndrome in severe COVID-19: interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. International Journal of Antimicrobial Agents. 2020;55(5):105954. doi:10.1016/j.ijantimicag.2020.105954
6.02. Grimble RF. Nutritional modulation of cytokine biology. Nutrition. 1998;14(7):634-640. doi:10.1016/S0899-9007(98)00010-0
6.03. Cavalli G, De Luca G, Campochiaro C, et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. The Lancet Rheumatology. 2020;2(6):e325-e331. doi:10.1016/S2665-9913(20)30127-2
7.01. Guo W, Li M, Dong Y, et al. Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes/Metabolism Research and Reviews. n/a(n/a):e3319. doi:10.1002/dmrr.3319
7.02. Lighter J, Phillips M, Hochman S, et al. Obesity in patients younger than 60 years is a risk factor for Covid-19 hospital admission. Clin Infect Dis. Published online April 9, 2020. doi:10.1093/cid/ciaa415
7.03. Simonnet A, Chetboun M, Poissy J, et al. High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obesity. n/a(n/a). doi:10.1002/oby.22831
7.04. Norris AL, Steinberger J, Steffen LM, Metzig AM, Schwarzenberg SJ, Kelly AS. Circulating Oxidized LDL and Inflammation in Extreme Pediatric Obesity. Obesity. 2011;19(7):1415-1419. doi:10.1038/oby.2011.21
7.05. Li S, Kievit P, Robertson A-K, et al. Targeting oxidized LDL improves insulin sensitivity and immune cell function in obese Rhesus macaques. Molecular Metabolism. 2013;2(3):256-269. doi:10.1016/j.molmet.2013.06.001
7.06. Nigam P, Bhatt S, Misra A, et al. Effect of a 6-Month Intervention with Cooking Oils Containing a High Concentration of Monounsaturated Fatty Acids (Olive and Canola Oils) Compared with Control Oil in Male Asian Indians with Nonalcoholic Fatty Liver Disease. Diabetes Technology & Therapeutics. 2014;16(4):255-261. doi:10.1089/dia.2013.0178
7.07. Maciejewska D, Ossowski P, Drozd A, et al. Metabolites of arachidonic acid and linoleic acid in early stages of non-alcoholic fatty liver disease—A pilot study. Prostaglandins & Other Lipid Mediators. 2015;121:184-189. doi:10.1016/j.prostaglandins.2015.09.003
7.08. Name M a. V, Chick JM, Savoye M, et al. 772-P: Effect of a Low n6/n3 PUFA Diet on Intrahepatic Fat Content in Obese Adolescents. Diabetes. 2019;68(Supplement 1). doi:10.2337/db19-772-P
7.09. Couillard C, Ruel G, Archer WR, et al. Circulating Levels of Oxidative Stress Markers and Endothelial Adhesion Molecules in Men with Abdominal Obesity. J Clin Endocrinol Metab. 2005;90(12):6454-6459. doi:10.1210/jc.2004-2438
7.10. Trpkovic A, Resanovic I, Stanimirovic J, et al. Oxidized low-density lipoprotein as a biomarker of cardiovascular diseases. Critical Reviews in Clinical Laboratory Sciences. 2015;52(2):70-85. doi:10.3109/10408363.2014.992063
7.11. Serbulea V, Upchurch CM, Schappe MS, et al. Macrophage phenotype and bioenergetics are controlled by oxidized phospholipids identified in lean and obese adipose tissue. PNAS. 2018;115(27):E6254-E6263. doi:10.1073/pnas.1800544115
8.01. Blasbalg TL, Hibbeln JR, Ramsden CE, Majchrzak SF, Rawlings RR. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am J Clin Nutr. 2011;93(5):950-962. doi:10.3945/ajcn.110.006643
8.02. Guyenet SJ, Carlson SE. Increase in Adipose Tissue Linoleic Acid of US Adults in the Last Half Century12. Adv Nutr. 2015;6(6):660-664. doi:10.3945/an.115.009944
8.03. Baker EH, Wood DM, Brennan AL, Clark N, Baines DL, Philips BJ. Hyperglycaemia and pulmonary infection. Proceedings of the Nutrition Society. 2006;65(3):227-235. doi:10.1079/PNS2006499
I thought it was good vitamin D levels that kept me in respiratory good health but maybe this is another factor I am on the right side of.